 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic microporous polymer networks by polymerisation of weakly coordinating cations with CO2-storage ability†
Sabrina
Fischer
a,
Aneta
Schimanowitz
a,
Robert
Dawson
*a,
Irena
Senkovska
b,
Stefan
Kaskel
b and
Arne
Thomas
*a
aTechnische Universität Berlin, Functional Materials, Hardenbergstr. 40, Berlin 10623, Germany. E-mail: arne.thomas@tu-berlin.de; rdawson@mailbox.tu-berlin.de
bTechnische Universität Dresden, Anorganische Chemie I, Bergstr. 66, Dresden 01062, Germany
First published on 29th May 2014
Abstract
Microporous organic polymer networks with weakly coordinating cations in their backbone have been synthesised by metal catalysed C–C bond forming reactions. A functionalised tetraphenylphosphonium ion was synthesised and successfully used as a tecton in a co-polymerisation with tetrakis(4-bromophenyl) methane using nickel catalysed Yamamoto coupling and with triethynylbenzene in a palladium catalysed Songashira–Hagihara reaction. The microporous materials showed an apparent BET surface area of 1455 m2 g−1 and 540 m2 g−1, respectively. The Yamamoto product provide a CO2 uptake of 2.49 mmol g−1 at 273 K and 1 bar. After ion exchange with chloride CO2 uptake is further increased to 2.85 mmol g−1.
Introduction
Microporous polymer networks are of increasing interest regarding their potential applications e.g. in the areas of separation, catalysis and gas storage.1–7 Several classes of these materials, which are built by polymerisation of rigid, two or three dimensional organic tectons have been described such as polymers of intrinsic microporosity (PIMs),8 covalent organic frameworks (COFs, CTFs)9–11 and conjugated microporous polymers (CMPs).12,13 Depending on the functional groups of the tecton different polymerisation reactions have been described to build such networks, such as oxidative polymerisations,5,14 polycondensations15 and trimerisations16,17 as well as Sonogashira,12,18,19 Suzuki20,21 or Yamamoto22,23 metal-catalysed C–C coupling reactions.Some microporous polymer networks show exceptionally high surface areas which allow the storage of high amounts of gases such as hydrogen or carbon dioxide.19,24,25 For example tectons based on tetraphenylmethane subunits exhibit BET surface areas of up to 5640 m2 g−1.23 Related tetrahedral tectons were reported, using silicon or germanium as central atom to connect the four phenyl rings.26–31 Beside group 14 elements, the exchange of the central carbon with elements from group 13 or 15 should yield negatively or positively charged tectons. The first group reporting a charged microporous polymer network based on such quaternary tectons were Zhang et al.,32 who used the positively charged phosphorus analogue of tetraphenylmethane, i.e. tetraphenylphosphonium cations with halides as counter ions. By polymerising this tecton via Yamamoto C–C coupling reaction a polymer was obtained that contained a mixture of phosphonium and phosphine moieties within the polymer resulting from partial decomposition of the phosphonium tecton during polymerisation. Recently we reported the use of tetrakis(4-bromo, 2,3,5,6-tetrafluorophenyl)borate as a tecton to generate a microporous anionic borate network (ABN) by Sonogashira–Hagihara coupling with 1,3,5-triethynylbenzene as comonomer.33 The counter cations, located within the pore channels, are highly accessible and can be easily exchanged, comparable to the extra framework cations found in zeolites. Ion exchange followed by the introduction of a ligand yielded an immobilised charged organometallic complex which can be used as a catalyst in the aerobic oxidation of alkenes.
Results and discussion
Here, we present the synthesis of two charged co-polymer networks containing a positively charged tetrahedral monomer, i.e. tetraphenylphosphonium and a neutral comonomer. The first polymer network is in principle the cationic counterpart of the previously reported anionic microporous polymer network (ABN). Instead of the negatively charged tetrakis(4-bromo, 2,3,5,6-tetrafluorophenyl)borate, the positively charged tetrakis(4-bromophenyl)phosphonium bromide was used as a monomer in a Sonogashira–Hagihara coupling with 1,3,5-triethynylbenzene. The second polymer network is generated by a Yamamoto coupling of the tetrakis(4-bromophenyl)phosphonium bromide with tetraphenylmethane. As the direct coupling of tetrakis(4-bromophenyl)phosphonium bromide yields to substantial decomposition of the monomer as reported by Zhang32 we envisioned that the use of a mixture of charged and non-charged monomers might help to stabilise the charges within the network in the same way as for zeolites. Löwenstein's rule for zeolites states that it is impossible to build in two aluminum atoms side by side in a tetrahedral composite. Even though this rule cannot simply be applied to organic networks, it inspired us to dilute the charged tectons with neutral ones to prevent the decomposition which was observed by Zhang et. al.34 and also by our own experiment. Note that also for the negatively charged borate tectons, we observed partial decomposition when a direct coupling was attempted, while the addition of a neutral comonomer yielded pure borate networks.34The synthesis of a phosphonium monomer containing polymerisable bromine groups was undertaken by firstly reacting 1,4-dibromobenzene and phosphorus trichloride to yield tris(4-bromophenyl)phosphine. A fourth bromophenyl group was introduced via palladium catalysed coupling reaction to obtain tetrakis(4-bromophenyl)phosphonium bromide (see ESI†). This tetrahedral monomer was then able to participate in a nickel catalysed Yamamoto reaction together with the neutral comonomer tetra(4-bromophenyl)methane to produce an insoluble yellow network in 97% yield (Scheme 1). The resulting cationic tetraphenylphosphonium-tetraphenylmethane bromide network (CPN-1-Br) is solely composed of phosphorous in the form of cationic phosphonium. Infrared spectra of the resulting polymer network clearly indicates the disappearance of the characteristic arylbromide bond at 1065 cm−1 (Fig. S1).† Together with the high yield this points to the successful inclusion of both monomers into the network.
 | ||
| Scheme 1 Synthesis of the CPN-1-Br. | ||
A second charged phosphonium microporous polymer network (CPN-2-Br) was formed by the palladium catalysed Sonogashira–Hagihara cross-coupling of tetrakis(4-bromophenyl)phosphonium bromide with 1,3,5-triethynylbenzene using optimised reaction conditions used previously for the synthesis of the anionic charged network33 (Scheme 2). Again high yields of ∼100% and the disappearance of the characteristic arylbromide bond at 1065 cm−1 (Fig. S1†) point to the inclusion of both monomers.
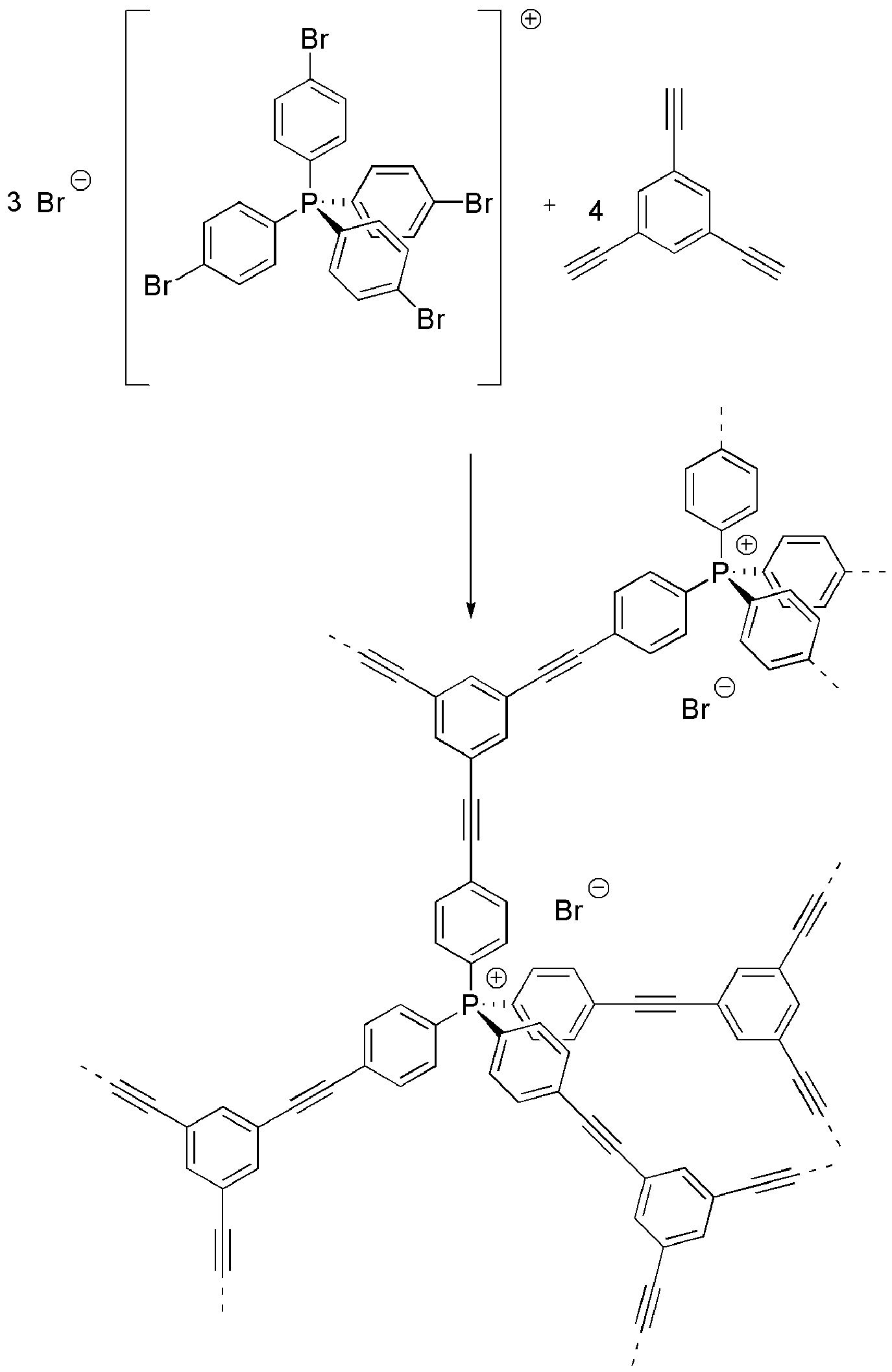 | ||
| Scheme 2 Synthesis of CPN-2-Br. | ||
The chemical identities of both materials were further confirmed by 13C- and 31P solid state NMR. For the Yamamoto linked network CPN-1-Br, signals at δ = 143, 138, 129, 125 and 63 ppm in the 13C {1H} MAS NMR indicate the presence of five different carbon environments (Fig. 1a). The signal at 63 ppm can be assigned to the central quaternary C-atom connected to four phenyl groups. The intense signal at 125 ppm can be assigned to the CAr–H of the tetraphenylphosphonium unit and the signal at 129 ppm can be assigned to the CAr–H of the tetraphenylmethane unit. The signal at 137 ppm corresponds to CAr–CAr–atoms as well as the carbons connected to phosphonium, while the aromatic carbons adjacent to the central carbons in the tetraphenylmethane unit exhibits a chemical shift of 143 ppm.
 | ||
| Fig. 1 a) 13C-NMR and (b) 31P-NMR spectra of CPN-1-Br. | ||
Fig. 2a shows the 13C {1H} MAS NMR of CPN-2-Br. The resonance at 89 ppm can be assigned to the acetylene groups and the signal at 122 ppm to the aromatic carbons of both monomers next to the triple bond. The intense signals at 126 ppm and 129 ppm can be assigned to the CAr–H of the tetraphenylphosphonium unit as well as of the 1,3,5-triethynylbenzene. The signal of the CAr–P–atoms is observed as a shoulder of the broad signal in the area between 133 ppm to 143 ppm. Notably, the 31P-NMR spectra of both materials (Fig. 1b and 2b) show just one intense signal at 24 and 25 ppm with a comparable shift to that seen for the monomer, tetrakis(4-bromophenyl)phosphonium bromide. No resonance due the cleaved corresponding phosphine can be observed in contrast to the network previously reported.32 We conclude therefore that the additional non-charged monomers were able to stabilise the charged phosphonium units in the polymer network.
 | ||
| Fig. 2 a) 13C-ssNMR and (b) 31P-ssNMR of CPN-Br. | ||
The networks were also characterised by nitrogen gas adsorption/desorption at 77 K. The Yamamoto network CPN-1-Br showed a Type I isotherm with a significant amount of gas adsorbed at low pressures followed by a large adsorption at higher pressures attributed to gas condensing into the voids between the polymer particles (Fig. 3). As often observed for the relatively soft polymer networks, the desorption branch does not close on the adsorption at low pressures.34 An apparent BET surface area of 1455 m2 g−1 was calculated for the network over a relative pressure range of 0.01–0.15 P/P0 (see Fig. S2†). The total pore volume was calculated to be 1.0 cm3 g−1. The Sonogashira network CPN-2-Br shows a lower BET surface area of 540 m2 g−1 compared to CPN-1-Br. A pore volume of 0.33 cm3 g−1 was calculated for CPN-Br.
 | ||
| Fig. 3 Nitrogen adsorption (closed)/desorption (open) isotherms at 77 K for CPN-1-Br (black squares), CPN-1-Cl (red circles) and CPN-2-Br (blue triangles). | ||
As previously shown for borate networks33 the counter ions can be exchanged after synthesis. Here, the as synthesised bromine ions can be simply and completely exchanged by chloride ions (CPN-1-Cl), by stirring the CPN-1-Br in saturated sodium chloride solution and methanol as shown by EDX analysis (see Fig. S11 and S12†). After ion exchange the BET surface area slightly increased to 1504 m2 g−1 with a pore volume of 1.0 cm3 g−1. Pore size distributions were calculated for the networks and showed the largest pores for CPN-1-Br and CPN-1-Cl to be centred around 16 Å (see Fig. S5†) similar to that for PPN-6.35 CPN-2-Br had a smaller pore size (7 Å).
The CO2 uptakes for the networks were measured at 273 (Fig. 4) and 195 K (Fig. S8 and S9) up to a pressure of 1 bar. It was assumed that for CO2 with its quadrupole moment, the introduction of network charges might have a considerable influence on the adsorption capacity. Furthermore, it was interesting to investigate if ion exchange within such networks influences the sorption characteristics, e.g. due to different size or Lewis acidity of the respective counter anions. CPN-1-Br showed an uptake at 273 K of 2.49 mmol g−1 while the network after anion exchange, CPN-1-Cl, showed an increased CO2 uptake of 2.85 mmol g−1 at 273 K and 1 bar. In comparison to the non-charged network (PAF-1/PPN-6) the CO2 uptake is increased by around 40% despite a much lower surface area (1504 m2 g−1vs. 5600 m2 g−1).25 This increased uptake can thus be attributed to the charges within the network. The Sonogashira network (CPN-2-Br) showed a lower uptake than the Yamamoto networks (1.55 mmol g−1) in line with its lower surface area. Using the two adsorption isotherms at 195 and 273 K the isosteric heats of adsorption were calculated using Clausius–Clapeyron equation (Fig. S10†). The heat of adsorption of CPN-1-Br at zero coverage was calculated to be 31.0 kJ mol−1 placing it amongst the highest reported microporous polymer networks and almost double the non-charged PAF-1/PPN-6 analogue (15.6–17 kJ mol−1).23,35 This value is similar to the acid containing PPN-6-SO3H (ref. 35) and CMP-1-COOH (ref. 36) networks, although lower than the amine functionalised PPN-6 series (such as PPN-6-DETA).37 The chloride exchanged network, showed a similar heat of adsorption while the Sonogashira network (CPN-2-Br) was slightly lower at 28.2 kJ mol−1 at zero coverage rising to 31.8 kJ mol−1 at a coverage of 7 cm3 g−1.
 | ||
| Fig. 4 Carbon dioxide adsorption (closed)/desorption (open) isotherms for CPN-1-Br (black squares), CPN-1-Cl (red circles) and CPN-2-Br (blue triangles) measured at 273 K. | ||
Conclusion
In conclusion, we have successfully synthesised two different charged microporous organic polymer networks CPN-1-Br and CPN-2-Br using tetrakis(4-bromophenyl)phosphonium bromide as a tecton. The tecton was stabilised by an analogous methane monomer which coupled trough a Yamamoto reaction with the tecton to form the CPMN-Br. The polymer network CPN-Br was obtained through a Sonogashira–Hagihara coupling with 1,3,5-triethynylbenzene. In this case 1,3,5-triethynylbenzene is the neutral, uncharged stabilising factor, which ensures the intactness of the tetrahedral phosphorus building unit during the reaction. Nitrogen sorption revealed a high surface area that, together with the charged nature of the polymer, ensured very good CO2 uptakes for the CPN-1-Br and CPN-1-Cl in comparison to non-charged network (PAF-1/PPN-6).Acknowledgements
We acknowledge the European Research Council for financial support within the project 278593_ORGZEO. The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007–2013) under REA grant agreement no. PIEF-GA-2011-300534.References
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed.
- H. J. Mackintosh, P. M. Budd and N. B. McKeown, J. Mater. Chem., 2008, 18, 573–578 RSC.
- J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702–705 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem.–Eur. J., 2005, 11, 2610–2620 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- J. Xia, S. Yuan, Z. Wang, S. Kirklin, B. Dorney, D.-J. Liu and L. Yu, Macromolecules, 2010, 43, 3325–3330 CrossRef CAS.
- R. S. Sprick, A. Thomas and U. Scherf, Polym. Chem., 2010, 1, 283–285 RSC.
- S. Yuan, S. Kirklin, B. Dorney, D.-J. Liu and L. Yu, Macromolecules, 2009, 42, 1554–1559 CrossRef CAS.
- S. Yuan, B. Dorney, D. White, S. Kirklin, P. Zapol, L. Yu and D.-J. Liu, Chem. Commun., 2010, 46, 4547–4549 RSC.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed.
- L. Chen, Y. Honsho, S. Seki and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426–4429 CrossRef CAS.
- T. Ben, H. Ren, S. Q. Ma, D. P. Cao, J. H. Lan, X. F. Jing, W. C. Wang, J. Xu, F. Deng, J. M. Simmons, S. L. Qiu and G. S. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- E. Stöckel, X. F. Wu, A. Trewin, C. D. Wood, R. Clowes, N. L. Campbell, J. T. A. Jones, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Chem. Commun., 2009, 212–214 RSC.
- R. Dawson, A. I. Cooper and D. J. Adams, Polym. Int., 2013, 62, 345–352 CrossRef CAS.
- M. Rose, W. Bohlmann, M. Sabo and S. Kaskel, Chem. Commun., 2008, 2462–2464 RSC.
- Y. Liao, M. Baskett, P. M. Lahti and F. Palacio, Chem. Commun., 2002, 252–253 RSC.
- D. Yuan, W. Lu, D. Zhao and H.-C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS PubMed.
- J. Fritsch, M. Rose, P. Wollmann, W. Böhlmann and S. Kaskel, Materials, 2010, 3, 2447–2462 CrossRef CAS PubMed.
- M. Rose, N. Klein, W. Bohlmann, B. Bohringer, S. Fichtner and S. Kaskel, Soft Matter, 2010, 6, 3918–3923 RSC.
- M. Rose, A. Notzon, M. Heitbaum, G. Nickerl, S. Paasch, E. Brunner, F. Glorius and S. Kaskel, Chem. Commun., 2011, 47, 4814–4816 RSC.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef CAS.
- S. Fischer, J. Schmidt, P. Strauch and A. Thomas, Angew. Chem., Int. Ed., 2013, 52, 12174–12178 CrossRef CAS PubMed.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880–2885 CrossRef CAS.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H.-C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173–1177 RSC.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta02022g |
| This journal is © The Royal Society of Chemistry 2014 |