 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidation of the reaction mechanism upon lithiation and delithiation of Cu0.5TiOPO4†
Peter
Bleith
a,
Maxence
Valla
b,
Petr
Novák
a and
Claire
Villevieille
*a
aPaul Scherrer Institut, Electrochemistry Laboratory, CH-5232 PSI Villigen, Switzerland. E-mail: Claire.Villevieille@psi.ch; Fax: +41 56 310 2688; Tel: +41 56 310 2410
bETH Zürich, Laboratory of Inorganic Chemistry, Vladimir-Prelog-Weg 1, CH-8093 Zürich, Switzerland
First published on 4th June 2014
Abstract
The reaction mechanism of Cu0.5TiOPO4 upon lithiation and delithiation was elucidated by XAS, 31P-NMR, XRD, EDX, and electrochemical methods. The material reacts with a combined insertion and conversion process, in which first copper is extruded irreversibly by forming LiTiOPO4. Afterwards, Ti4+ is reduced reversibly in an insertion reaction followed by a conversion reaction. The conversion reaction leads to amorphization of the sample while titanium is reduced to oxidation states below 2+.
Introduction
Lithium-ion batteries are one of the most used electrochemical energy storage systems nowadays. In order to further improve the system, there is a constant search for new electrode materials, allowing a higher voltage or increasing the capacity of the cells. Some materials exhibit a high specific charge but react at potentials that are not useful in a full cell because using them results in less energy stored in the system. However, understanding such a reaction mechanism is crucial in the way of designing new materials with high specific charge reacting at a reasonable potential.Metal titanium oxyphosphates, also called metal titanyl phosphates, (M0.5TiOPO4 with M = Ni2+, Fe2+, Co2+, Cu2+, and Mg2+) are a family of isostructural materials. In 2005, it was reported that Ni0.5TiOPO4 exhibits a quite high specific charge of 415 A h kg−1 which corresponds to a reaction with ≈3 Li+ per formula unit (f. u.) upon lithiation to 0.5 V vs. Li+/Li and ≈2 Li+ per f. u. upon delithiation to 4.0 V.1
In the following years, studies were ongoing to understand the reason for the high specific charge of Li2xNi0.5−xTiOPO4,2–7 Co0.5TiOPO4,8 and Fe0.5TiOPO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 but none of these completely elucidated the reaction mechanism. For example, it is unclear at which potential and to which degree the M-atom is reduced upon lithiation and re-oxidized upon delithiation in different M0.5TiOPO4: Belharouak and Amine claimed evaluating the Ni K-edge that Ni2+ remains unchanged1 but Hollmark et al. claimed by observing the Ni L-edge that Ni2+ is reduced upon lithiation to an oxidation state “somewhat below Ni1+” and is not completely re-oxidised to Ni2+ upon delithiation.2 Essehli et al. did not detect any metallic Co upon lithiation using XRD10 whereas Lasri et al. detected metallic Fe upon chemical lithiation of Fe0.5TiOPO4 using Mössbauer spectroscopy.9 However, all studies could not explain the source for the specific charge of this family of materials.
9 but none of these completely elucidated the reaction mechanism. For example, it is unclear at which potential and to which degree the M-atom is reduced upon lithiation and re-oxidized upon delithiation in different M0.5TiOPO4: Belharouak and Amine claimed evaluating the Ni K-edge that Ni2+ remains unchanged1 but Hollmark et al. claimed by observing the Ni L-edge that Ni2+ is reduced upon lithiation to an oxidation state “somewhat below Ni1+” and is not completely re-oxidised to Ni2+ upon delithiation.2 Essehli et al. did not detect any metallic Co upon lithiation using XRD10 whereas Lasri et al. detected metallic Fe upon chemical lithiation of Fe0.5TiOPO4 using Mössbauer spectroscopy.9 However, all studies could not explain the source for the specific charge of this family of materials.
In this manuscript we elucidate the reaction mechanism of Cu0.5TiOPO4 by combining electrochemical information with information from XRD, XAS, NMR, and EDX.
Experimental
Cu0.5TiOPO4 was prepared by a co-precipitation method published recently.11 The synthesis results in micron-sized particles of both polymorphs, α- and β-Cu0.5TiOPO4 with some percentage of Cu3(PO4)2 as impurities.For electrochemical studies (at 25 ± 0.1 °C), the working electrodes were prepared in three different ways: (1) a 9:1 mixture of the powders of Cu0.5TiOPO4 and Super-P carbon, (2) a self-standing film or (3) a dried slurry doctor-bladed on a copper-foil current collector. Both, the film and the slurry consisted of the Cu0.5TiOPO4 active material, Super-P carbon conductive additive, and Kynarflex® 2801 binder with a composition of 70:10:20 wt% for the film and 80:10:10 wt% for the slurry, respectively. Coin-like electrochemical cells with a lithium metal counter electrode, a glass fiber separator, and a 1:1 mixture of ethylene carbonate and dimethyl carbonate with 1 M LiPF6 as the electrolyte were assembled in an argon-filled glove box. The film was used for in situ XRD and EDX measurements. The electrodes on copper-foil were used for the GITT and NMR measurements. If not stated otherwise a specific current of 42.2 A kg−1 with respect to the active material was used. For the GITT measurement, the current was applied for 20 min followed by a relaxation period of 10 h. Ex situ samples for XRD and XAS measurements were cycled with a current density of 14.07 A kg−1 and for NMR measurements at 4.22 A kg−1.
X-ray diffraction (XRD) measurements were performed at room temperature with a PANalytical Empyrean diffractometer using copper or molybdenum Kα-radiation. In situ measurements were performed in a cell with a beryllium window in Bragg-Brentano geometry. Ex situ samples were measured in capillaries.
X-ray absorption spectroscopy (XAS) measurements were performed at the SuperXAS beamline at SLS (PSI, Villigen, Switzerland). In an argon-filled glove box, the cycled powder was filled in capillaries (d = 0.7 mm) and sealed. As reference samples, CuO, Cu2O, TiO2 (rutile), Li4Ti5O12, Ti2O3, and TiO were used. Except for Ti2O3 and TiO, pellets of the reference powder mixed with cellulose were pressed and measured. The other two reference samples were measured undiluted in capillaries. Fluorescence spectra were recorded at the titanium K-edge (except for the titanium foil reference which was measured in transmission) and transmission spectra were recorded at the copper K-edge. For the analysis of the data, the software-package Demeter was used.12 For all data, the background was subtracted and the energy of E0 was set to a fraction (50%) of the edge step.
For NMR measurements, cells were disassembled in an argon-filled glove box and the electrodes were washed with DMC. Afterwards the powder was scratched from the copper current collector, ground in a mortar, and filled in a 4 mm solid state NMR rotor. All the spectra were acquired using a 400 MHz (9.4 T) ultrashield Bruker spectrometer equipped with a 4 mm HX MAS probe. 31P single excitation with an Rf field of 125 kHz and a 84745 Hz spectral width was used to record the spectra.
For EDX measurements, cells were disassembled in an argon-filled glove box. The lithium electrodes were washed in DMC and afterwards placed on an SEM holder. The sample was transferred under vacuum to the SEM (Ultra55 by Carl Zeiss).
Results and discussion
GITT measurements can give indications on the type of reaction of a material upon lithiation and delithiation; with small overpotentials the reaction is most likely an insertion reaction, with high overpotentials the reaction is most likely a conversion reaction. Fig. 1 shows a GITT measurement of an electrode with Cu0.5TiOPO4 in the first cycle. The lithiation can be separated into four parts corresponding to the amount of Li+ per f. u. reacted: (0–1 Li+) high overpotential, (1–2 Li+) low overpotential, (2–3 Li+) high overpotential, and (>3 Li+) medium overpotential. From these data it can be assumed that the reaction with the first Li+ per f. u. is a conversion or extrusion reaction, while the second one is an insertion reaction, followed by a conversion reaction.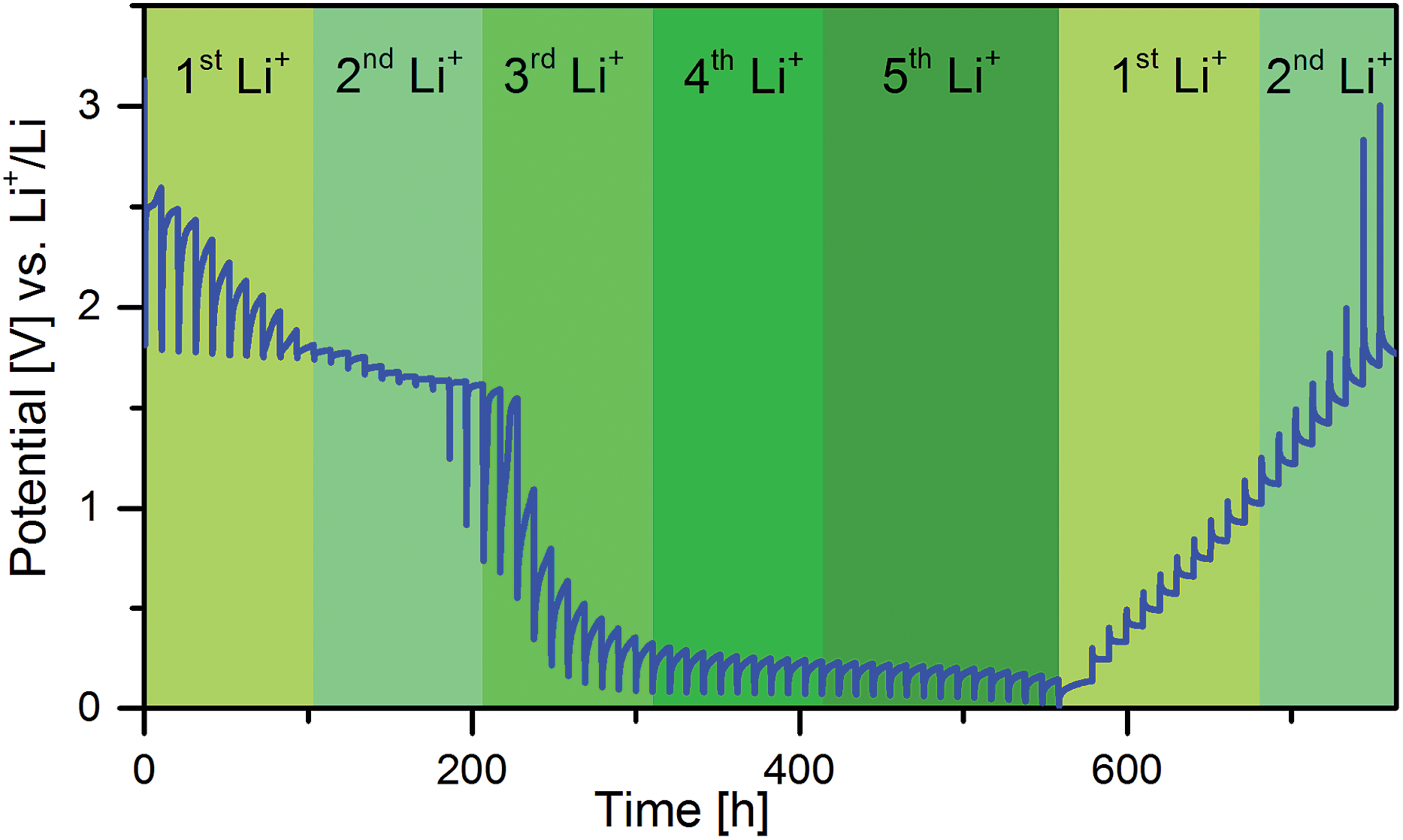 | ||
| Fig. 1 GITT-measurement during the first cycle of Cu0.5TiOPO4. | ||
The evolution of phases occurring during cycling between 3.0 V and 1.0 V for the first cycle and between 3.0 V and 10 mV during the second cycle was followed by in situ XRD (Fig. 2). At the end of the first plateau after a reaction with approx. 1 Li+ per f. u., reflections of Cu0.5TiOPO4 change to a similar phase and at the position of the copper 111-reflection (43.3°) a broad peak appears. Upon further cycling to 1.0 V (≈2 Li+ per f. u.), the reflection of the copper gets more pronounced and the phase similar to Cu0.5TiOPO4 changes slightly again. This change is reversible upon delithiation to 3.0 V. The following lithiation to 10 mV leads to amorphisation of the structure, with the exception of copper. Upon following delithiation, the copper peak shifts slightly to higher angles (to 43.45°).
 | ||
| Fig. 2 Contour plot of an in situ XRD measurement at a specific current of 16.9 A kg−1 (blue: low intensity; red: high intensity). On the right hand side, the corresponding galvanostatic curve is shown. | ||
In order to get a clearer view on the reaction mechanism, samples were cycled to key positions in the galvanostatic curve and ex situ XRD measurements were performed on these samples (Fig. 3). After the lithiation with 1 Li+ per f. u. (sample c), two main phases can be detected, namely (triclinic) LiTiOPO4 and metallic copper (for the refinement see ESI, Fig. S1†). Reflections around 10° are most likely due to remainders of the electrolyte salt. Upon the reaction with the second Li+ per f. u. (sample e), a similar phase to LiTiOPO4 is formed. This phase was not yet reported in the literature. Delithiation to 3.0 V (sample g) leads back to LiTiOPO4. Lithiation to more negative potentials leads, as for the in situ measurements, to amorphization of the material (sample h) indicating a conversion type reaction which is not reversible (sample k). Upon lithiation to 10 mV the reflections of copper do not change significantly; upon delithiation to 3.0 V they shift slightly to higher angles (from 19.5 to 19.6°). This shift of the copper reflection is reversible (ESI, Fig. S2†).
 | ||
| Fig. 3 (a) Ex situ XRD measurements of Cu0.5TiOPO4 cycled to key points in the first cycle. (b) shows the corresponding galvanostatic curve. | ||
The reason for the shift of the copper reflection is unclear yet. Two hypotheses could explain this behavior: (1) copper is extruded as small particles (crystallite size ≈6 nm from an analysis based on the Scherrer-equation). These particles are stuck at phase boundaries between grains of the now amorphous LiTiOPO4. Delithiation might lead to an expansion of those grains which then compress the copper particles in between. (2) At elevated temperatures, an existence of a solid solution of lithium in copper with up to 20 at% lithium is possible.13 This could possibly happen to nanoparticles as well. The change of the lattice constant reported by Klemm and Volavšek13 matches with the change we observed. The weak scattering of X-rays by lithium and the small change in the lattice constant might be the reason why this reaction is not seen in the EXAFS data. Note that with the techniques we have at hand, it is not possible to distinguish between both hypotheses. Though we think that (2) is more likely.
The extrusion of copper can also be confirmed by EXAFS measurements at the copper K-edge performed on the same samples of the ex situ XRD measurements. As shown in Fig. 4, Cu0.5TiOPO4 lithiated to 1.78 V (before the first plateau) is similar to the starting material while all other samples (starting from the sample c, which was lithiated to 1.71 V (≈1 Li+ per f. u.) up to sample g, which was delithiated to 3.0 V) show a similar spectrum to a metallic copper foil. This indicates that copper is irreversibly extruded from Cu0.5TiOPO4 during the reaction with the first Li+ per f. u. and is extruded as agglomerates of metallic copper. This is also supported by the fitting of the data with the possible scattering pathways (ESI, Fig. S3 and Table S1†). While the pristine sample and the sample cycled to 1.78 V could be fitted with α-Cu0.5TiOPO4, the sample cycled to 1.71 V could only be fitted with metallic copper.
 | ||
| Fig. 4 Fourier transform of the recorded EXAFS-spectra of cycled samples of Cu0.5TiOPO4 (for labeling see Fig. 6). | ||
In order to identify the reacting atoms responsible for the different plateaus, ex situ XAS spectra at the titanium and copper K-edges were recorded from samples cycled to key points in the first cycle (Fig. 5). Fig. 6 shows the change of the copper and titanium absorption edges upon lithiation and delithiation of Cu0.5TiOPO4. The analysis of this change gives another evidence that copper is irreversibly reduced from Cu2+ to Cu0 between 1.78 and 1.71 V upon lithiation (between samples b and c). The analysis of the titanium K-edge shows that titanium is reversibly reduced in the plateau between 1.71 and 1.0 V (samples c to d) from Ti4+ to Ti3+. Upon further lithiation from 0.5 to 0.01 V (samples e to h), titanium is reduced further to an oxidation state below 2+. In both cases, upon cycling to 0.01 V and 0.5 V, the reduction of the titanium is reversible; upon delithiation to 3.0 V, it is oxidized to Ti4+ again.
 | ||
| Fig. 5 Selected normalized XAS spectra at (a) the titanium and (b) the copper K-edge (for labeling see Fig. 6). | ||
 | ||
| Fig. 6 Change of the energy of the copper- and the titanium absorption edge at half of the edge step of ex situ samples of Cu0.5TiOPO4. The position of the absorption edge of the measured reference materials is shown as well. At the bottom, the corresponding galvanostatic curve is plotted. | ||
To which oxidation state titanium was reduced upon lithiation to 0.01 V is not completely clear by the XANES data due to two reasons: (1) the position of the absorption edge can not only shift due to a reduction of a species but also due to changes in the coordination of the sample. Since at this stage of the cycling the sample turns amorphous, there are certainly major structural changes. If these changes affect the coordination of the titanium, this cannot be distinguished from a change in the oxidation state using XANES measurements. Calculations by “materialsproject.org”14,15 show for the isostructural Ni0.5TiOPO4 and Fe0.5TiOPO4 that thermodynamically both titanium and the phosphate group can be reduced upon lithiation to 0.01 V. (2) Relaxation effects could be the consequence of a partial re-oxidation of the ex situ sample, e.g. by a reaction with other components present in the cell like electrolyte and/or the SEI.
Ex situ 31P MAS NMR measurements can help to elucidate whether phosphate is reduced upon lithiation to 0.01 V or not. Fig. 7a shows the 31P NMR spectrum of Cu0.5TiOPO4 as synthesized; due to the paramagnetism of Cu2+, the chemical shift range is larger than usual 31P NMR, starting from −200 ppm to +500 ppm. We can distinguish two peaks, one being at 429 ppm assigned to the phosphorus nucleus in Cu0.5TiOPO4 and one at 28 ppm assigned to Cu3(PO4)2 (impurity detected by XRD in the as-synthesized compound). Starting from this material, NMR spectra were acquired at different cycled points in the galvanostatic curve (Fig. 7(a)–(e)). At the last point, where the potential is back to 3.0 V and therefore at the end of the cycle, one single peak is observed (Fig. 7e) at 11 ppm assigned to Li3PO4 (according to the measurement of a commercial powder, Fig. 7f) meaning that the initial Ti3+ is reduced and is not part of the final 31P compound.
 | ||
| Fig. 7 31P MAS NMR spectra of ex situ samples of Cu0.5TiOPO4 and pure Li3PO4. The top spectrum is an addition of two spectra acquired at two different transmitter positions (0 ppm and 250 ppm) to cover a larger window width. For all the spectra the number of scans was 512 K and the recycling delay was set to 1 s. | ||
In the fully lithiated state at 0.01 V, one single peak is observed (Fig. 7d) at 11 ppm assigned to Li3PO4 (Fig. 7f) meaning that the phosphate group was not reduced at these potentials. The Li3PO4 does not change upon delithiation to 3.0 V (Fig. 7e). Note that when the irreversible reduction of Cu2+ to Cu0 happens, a 31P containing intermediate is formed (Fig. 7b). Two sharp peaks at respectively −3 and −9 ppm appeared at a potential of 1.75 V and disappear below 1.5 V. Those two peaks are assigned to LiTiOPO4 in the triclinic phase. To confirm this hypothesis NMR CASTEP16 calculations were carried out on the LiTiOPO4 in the triclinic and orthorhombic phase. The results were referenced with respect to triphenyl phosphine (ESI, Fig. S3†). The triclinic phase gave two signals (5 ppm apart from each other) with equal intensity while the orthorhombic gave only one signal at different chemical shifts (ESI, Table S2†). Experimentally the two peaks are separated by 6 ppm and have the same intensities. This confirms the presence of the triclinic phase at a potential of 1.75 V.
This excludes the possibility that phosphate is reduced upon lithiation to 0.01 V, leaving only two further components in the electrochemical system to be reduced between 0.5 and 0.01 V: the conductive additive Super-P and Ti3+. Additional experiments, with electrodes that contained only Super-P carbon as the active material, showed that Super-P carbon exhibits a specific charge of 574 A h kg−1 in the first lithiation (including the formation of an SEI). Based on the 10% content of Super-P in the electrode, the total reaction of Cu0.5TiOPO4 (≈5.4 Li+ per f. u.) is thus by <0.5 Li+ per f. u. less than it seems to be, and cannot explain the reaction with ≈3 Li+ per f. u. between 0.5 and 0.01 V. This indicates together with the results from XANES that titanium is reduced from Ti3+ at these potentials at least to Ti2+, most likely further. The rest of the specific charge could be explained by the reduction of the electrolyte on newly exposed surfaces, i.e. SEI formation type reactions.
The irreversible nature of the reduction of Cu2+ to Cu0 upon cycling at potentials negative to 3.0 V is obvious because metallic copper is not noticeably oxidized at these potentials (e.g. the copper current collector). Experiments on self-standing films of Cu0.5TiOPO4 without a copper current collector show that the reduced copper can be re-oxidized electrochemically (Fig. 8). However, the re-oxidation is not complete and the material shows a very strong capacity fading (almost no reaction detectible after 3 cycles).
 | ||
| Fig. 8 Galvanostatic curve of Cu0.5TiOPO4 cycled 3 times between 1.7 V and 4.0 V with a specific current of 14.06 A kg−1. | ||
The explanation for this behavior can be found by EDX measurements on the lithium counter-electrode of these cells (Fig. 9). Copper can be detected on lithium after cycling to 4.0 V after prior cycling to 1.7 V but not after solely cycling to 1.7 V. Since the only possible source of copper in the complete system was Cu0.5TiOPO4, the copper must be dissolved, probably as Cu2+, upon oxidation in the electrolyte and was then reduced on metallic lithium. Measurements of the counter electrode of an uncycled cell of Cu0.5TiOPO4 showed that also copper ions from the starting compound are dissolved at least partly in the electrolyte within two weeks and are then reduced at the lithium counter electrode. Note that impurities from Na, Mg, Al, K, and Ca might come from washing the lithium in a glass beaker with DMC.
 | ||
| Fig. 9 EDX spectra of lithium counter electrodes and working electrodes of Cu0.5TiOPO4 after cycling to 1.7 V, cycling 10 times between 1.7 V and 4.0 V, and an uncycled cell (all stored for two weeks after assembly). | ||
Conclusions
Based on the results above, a reaction mechanism is suggested as summarized in Table 1 where the different reaction steps of Cu0.5TiOPO4 upon lithiation and delithiation are shown. The extrusion and reduction of copper could be detected by XRD, XANES, and EXAFS. The re-oxidation of copper was shown indirectly by electrochemical methods combined with EDX. The reduction and oxidation of titanium was shown by XANES, taking into consideration the not changing PO43− measured by 31P MAS NMR. The reduction from Ti4+ to Ti3+ is an insertion reaction; further reductions take place in conversion processes.| Potential | Reaction | Type | ||
|---|---|---|---|---|
| a Potential and reaction type given for lithiation to 2 Li+ per f. u. | ||||
| Lithiation/reduction | 1st Li | 1.75 V | Cu2+ → Cu0 | Conversion (extrusion) |
| 2nd Li | 1.70 V | Ti4+ → Ti3+ | Insertion | |
| 3rd Li | ≈0.1 V | Ti3+ → Ti2+ | Conversion | |
| Delithiation/re-oxidation | 1st Li | <1.0 V | Ti3+ → Ti2+ | Conversion |
| 2nd Li | 1.75 Va | Ti4+ → Ti3+ | Insertiona | |
| 3rd Li | >3.5 V | Cu0 → Cu2+ (dissolved) | Conversion | |
Acknowledgements
Swiss National Science Foundation is thanked for financial support. Dr Olga Safonova from the SuperXAS beamline at SLS (Villigen, Switzerland) is thanked for the support during the measurements.References
- I. Belharouak and K. Amine, Electrochem. Commun., 2005, 7, 648 CrossRef CAS PubMed.
- H. M. Hollmark, K. Maher, I. Saadoune, T. Gustafsson, K. Edström and L. C. Duda, Phys. Chem. Chem. Phys., 2011, 13, 6544 RSC.
- K. Maher, K. Edström, I. Saadoune, T. Gustafsson and M. Mansori, J. Power Sources, 2011, 196, 2819 CrossRef CAS PubMed.
- V. A. Godbole, C. Villevieille, H.-H. Sommer, J.-F. Colin and P. Novák, Electrochim. Acta, 2012, 77, 244 CrossRef CAS PubMed.
- R. Essehli, B. El Bali, A. Faik, S. Benmokhtar, B. Manoun, Y. Zhang, X. J. Zhang, Z. Zhou and H. Fuess, J. Alloys Compd., 2012, 530, 178 CrossRef CAS PubMed.
- K. Lasri, M. Dahbi, A. Liivat, D. Brandell, K. Edström and I. Saadoune, J. Power Sources, 2013, 229, 265 CrossRef CAS PubMed.
- R. Eriksson, K. Maher, I. Saadoune, M. Mansori, T. Gustafsson and K. Edström, Solid State Ionics, 2012, 225, 547 CrossRef CAS PubMed.
- R. Essehli, B. El Bali, H. Ehrenberg, I. Svoboda, N. Bramnik and H. Fuess, Mater. Res. Bull., 2009, 44, 817 CrossRef CAS PubMed.
- K. Lasri, I. Saadoune, Y. Bentaleb, D. Mikhailova, H. Ehrenberg, L. Häggström and K. Edström, Solid State Ionics, 2012, 224, 15 CrossRef CAS PubMed.
- R. Essehli, B. E. Bali, H. Ehrenberg, I. Svoboda, N. Bramnik and H. Fuess, Mater. Res. Bull., 2009, 44, 817 CrossRef CAS PubMed.
- P. Bleith, P. Novák and C. Villevieille, J. Electrochem. Soc., 2013, 160, A1534 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- W. Klemm and B. Volavšek, Z. Anorg. Allg. Chem., 1958, 296, 184 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef PubMed.
- F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235121 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z Kristallogr, 2005, 220, 567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Refinement of the XRD of Cu0.5TiOPO4 cycled to 1.71 V (sample c). In situ XRD measurement of Cu0.5TiOPO4 cycled two times. Fit of the EXAFS data of Cu0.5TiOPO4 cycled to 1.78 V, 1.71 V, and uncycled (samples a–c). The 31P NMR spectrum of triphenyl phosphine. A table with calculated and experimental chemical shifts of triphenyl phosphine and LiTiOPO4. See DOI: 10.1039/c4ta01627k |
| This journal is © The Royal Society of Chemistry 2014 |