
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogels by supramolecular crosslinking of terpyridine end group functionalized 8-arm poly(ethylene glycol)†
Rong
Wang
a,
Mike
Geven
a,
Pieter J.
Dijkstra
a,
Penny
Martens
b and
Marcel
Karperien
*a
aMIRA Institute for Biomedical Technology and Technical Medicine, Department of Developmental Bioengineering, Faculty of Science and Technology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: h.b.j.karperien@utwente.nl; Tel: +31 (0)53 4893323
bUNSW Australia, Graduate School of Biomedical Engineering, Sydney, New South Wales 2052, Australia
First published on 4th August 2014
Abstract
Metallo supramolecular assemblies of an 8-arm poly(ethylene glycol) partially substituted with terpyridyl end-groups and the transition metal ions Ni2+, Fe2+, Co2+ and Zn2+ were studied for their nano-particle formation at dilute conditions and gelation at higher concentrations. The large differences in dissociation rate constants of the metal ligand complexes largely determine the assembly behavior. Thermodynamically stable complexes are generated with Ni2+ and Fe2+ chlorides, which lead to distinct particle sizes of ∼200 nm in dilute conditions. The Co2+ and Zn2+ chlorides provide multiple size distributions revealing that mono and bis-complexes are present at equilibrium. Upon complexation, terpyridyl groups move to the outer sphere giving aggregates with a charged surface. At polymer concentrations above 5 wt%, crosslinking upon addition of transition metal ions provides hydrogels. Elastic hydrogels were obtained with Ni2+, Fe2+ and Co2+ having storage moduli in excess of 20 kPa, whereas Zn2+ gels are relatively viscous. Only Zn2+ gels show a thermoreversible sol to gel transition at a temperature of 25 °C independent of polymer concentration.
Introduction
Polymer gels are networks of polymer molecules that are either covalently or physically crosslinked and expanded throughout their volume by a fluid.1 Physically cross-linked polymer gels are based on supramolecular chemistry, classically defined as the chemistry of complex formation through non-covalent interactions.2 These interactions include dipole–dipole interactions, ionic bonding, hydrogen bonding, π–π stacking or combinations of these, such as ion–dipole interactions.2,3 The properties of physically cross-linked gels may vary widely, especially when the interactions are governed by dynamic equilibrium processes. As an example, cation–anion interactions can yield bond strengths up to 350 kJ mol−1. This type of interaction has been used in several supramolecular polymer gels like calcium alginate gels,4,5 gellan gum6 and chitosan polyelectrolyte complexes.7 Although cation–anion interactions can yield mechanically strong gels, the cross-links in these gels can be dynamic due to the relatively high kinetic lability of the interaction.8 This latter can result in interesting features of the gels, such as gelling transitions effected by temperature, pH or ultrasound and the ability to self-heal.9–11Gels formed by supramolecular cross-linking using metal ions as guests and ligands as hosts will also strongly depend on the predominant equilibrium state. A gel is only formed when the host–guest complex is favored at equilibrium. Bond strengths in metal ligand complexes can be as high as those of covalent bonds.12 In certain cases highly stable complexes are formed when multiple Brønsted bases in a molecule donate electrons to a single Lewis acid which is known as the chelate effect.13,14 Importantly, although these interactions can have a high thermodynamic stability, the metal coordination bond can nonetheless possess a relatively high kinetic lability. This combination of relatively high thermodynamic bond stability and kinetic lability makes metal coordination bonds an interesting choice for use as cross-links in polymeric gels. Such systems are currently attracting increasing interest and are termed metallo-supramolecular polymer gels (MSPGs).15
Terpyridines are well known to effectively bind to transition metal ions, although reports of binding to alkali, alkaline earth and rare earth metal ions exist as well.16–19 Binding with third row transition metal ions ideally proceeds in a two-step process.20 The equilibria involve the stepwise coordination of two terpyridine moieties to a metal ion with different stability constants for the metal–mono(terpyridine) complex (K1) and metal–bis(terpyridine) complex (K2), respectively. The overall equilibrium is represented by the overall stability constant β, the product of K1 and K2. The stability constants are equilibrium constants and are defined by their association and dissociation rate constants.21 The stability of transition metal bis(terpyridine) complexes is affected by several factors, such as the metal ion, temperature, pH and solvent. The metal ion is of paramount importance in this respect as it is known that the overall stability constants (β) of transition metal ion–bis(terpyridine) complexes in water follows the series Ni2+ > Fe2+ > Co2+ > Zn2+.22,23
Several extended overviews on terpyridine chemistry and their complexes with a variety of transition metal ions have been published in the past decade.24,25 However, research on MSPGs has mainly focused on complex formation in organic solvents, i.e. on the formation of organogels and only minor research has been directed to hydrogels. Reports on MSPGs formed in water cover either terpyridyl end-group modified thermosensitive block copolymers, a terpyridyl grafted amphiphilic block copolymer and end-group modified 3 or 4 armed poly(ethylene glycol).26 Chiper et al. end modified commercially available pluronics with terpyridyl groups and determined their ability to form nano-particles in dilute solution upon complexation with NiCl2.27 The aqueous aggregation of these modified pluronics into micelles in the absence of a metal ion took place via hydrophobic interactions of the hydrophobic blocks and terpyridyl groups. Addition of NiCl2 led to the rearrangement and disruption of the micellar aggregates due to the hydrophilicity of the metal ion–terpyridine complexes generated. Furthermore, intramicellar bis(terpyridine) complexes were formed due to the relatively high effective concentration of these groups at the micelle coronas. At high concentrations of 20 wt%, the pluronic F127 end modified with terpyridyl groups in the presence of half an equivalent Ni2+ ions showed thermoreversible gelation behaviour with a transition temperature of 34 °C. As suggested, gelation depends on the packing of micelles and not on intermicellar Ni2+–terpyridine complexes formed.
Jochum et al. showed that a terpyridine modified block-copolymer of hydrophilic poly(triethyleneglycol methylether methacrylate) (PTEGMA) grafted with a low percentage of terpyridyl groups and polystyrene readily formed gels upon the addition of NiCl2.28 Compared to gels based on only terpyridine modified PTEGMA, the self-assembly of the polystyrene blocks led to additional cross-links resulting in an increased modulus of the gels.
Only a few papers on terpyridyl modified PEG's that can be crosslinked into MSPGs in an aqueous environment have appeared in literature. Schmatloch and Schubert reported on a low molecular weight three-arm PEG terminated with terpyridyl groups.26 When combined with a linear terpyridyl end modified PEO, the addition of Fe2+ ions it was shown that the branched structure acted as a chain stopper instead of crosslinker. Kimura prepared 4-armed PEG, terminated with terpyridyl groups and studied the complexation behavior of this polymer with Fe2+ in methanol. The dried polymer formed a stable gel in water at a concentration of 20 wt%. They also showed that varying the pH from 1 to 13 or heating to 90 °C did not give visible changes.29 A three armed terpyridyl terminated PEG was recently studied for gelation with cobalt ions. Hydrogels were only formed from a water-soluble polymer having a Mn of 8000, but not with lower molecular weight polymers.30 Recently, Yoshida and coworkers showed that terpyridyl end-modified 4 and 8 armed PEG form hydrogels upon complexation with Ru2+ ions. They used the complexes as a catalyst in the Belousov–Zhabotinsky reaction. The mechanical properties of the gels are almost independent on the number of arms, but gelation kinetics is higher for the 8-armed polymer.31
Although the above presented examples show that terpyridyl-modified polymers may be used for the design of hydrogels, only minor attention has been given to the assembly process and stability of these systems in an aqueous environment using different transition metal ions. In a recent paper by Rossow and Seiffert supramolecular hydrogels based on 4-arm PEG end-modified with terpyridyl end-groups and Mn2+, Zn2+ and Co2+ ions were described.32 They showed that network inhomogeneity is higher in Co2+ complexed gels compared to Zn2+ complexed gels. However, this nanometer-scale inhomogeneity seems to enforce rather than weaken the gel mechanical properties. In this paper, we describe supramolecular gels formed by the self-assembly of an 8-arm PEG end modified with terpyridyl groups via metal–ligand interaction using Ni2+, Fe2+, Co2+ and Zn2+ ions. The aggregation at dilute aqueous conditions and gelation properties at concentrations above 5 wt% were determined. Importantly, we show that the kinetic stability of the metal–ligand crosslinks in the gels determines the temperature dependent gelation properties of the hydrogels.
Experimental
Materials
8-arm poly(ethylene glycol) (8PEG, hexaglycerol core, Mw = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was acquired from Jenkem Technology (Allen, Texas, USA) and purified before use by dissolution in dichloromethane and precipitation in cold diethyl ether. The polymer was freeze dried overnight before use. Dimethyl sulfoxide (anhydrous, ≥99.9%), 4′-chloro-2,2′:6′,2′′-terpyridine (99%), potassium hydroxide (BioXtra, ≥85%), sodium sulfate (anhydrous, ACS reagent, ≥99%), cobalt(II) chloride (anhydrous, 99.999%), zinc chloride (anhydrous, ≥99.999%), nickel(II) chloride hexahydrate (99.999%) and iron(II) chloride (anhydrous, 99.99%) were purchased from Sigma-Aldrich (Zwijndrecht, the Netherlands) and were used as received. Dichloromethane (AR, stabilized by Amylene, ≥99.9%) and diethyl ether (AR, stabilized by BHT, ≥99.5%) were from Biosolve and chloroform (for analysis, ≥99.8%) was from Emsure and were used as received. Water used in this study was demineralized water purified by a Millipore Synergy system and bubbled with nitrogen gas for at least 30 minutes before use. Dialysis membranes (Spectra/Por molecular porous membrane tubing 4, MWCO: 12–14000) were acquired from Spectrum Laboratories, Inc. and were soaked in water for 30 minutes before use. Deuterated chloroform (99.8 atom% D, 0.5 wt% silver foil as stabilizer) was from Aldrich. Durapore PVDF membrane, 0.22 μm pore size filters were from Millex GV.
000) was acquired from Jenkem Technology (Allen, Texas, USA) and purified before use by dissolution in dichloromethane and precipitation in cold diethyl ether. The polymer was freeze dried overnight before use. Dimethyl sulfoxide (anhydrous, ≥99.9%), 4′-chloro-2,2′:6′,2′′-terpyridine (99%), potassium hydroxide (BioXtra, ≥85%), sodium sulfate (anhydrous, ACS reagent, ≥99%), cobalt(II) chloride (anhydrous, 99.999%), zinc chloride (anhydrous, ≥99.999%), nickel(II) chloride hexahydrate (99.999%) and iron(II) chloride (anhydrous, 99.99%) were purchased from Sigma-Aldrich (Zwijndrecht, the Netherlands) and were used as received. Dichloromethane (AR, stabilized by Amylene, ≥99.9%) and diethyl ether (AR, stabilized by BHT, ≥99.5%) were from Biosolve and chloroform (for analysis, ≥99.8%) was from Emsure and were used as received. Water used in this study was demineralized water purified by a Millipore Synergy system and bubbled with nitrogen gas for at least 30 minutes before use. Dialysis membranes (Spectra/Por molecular porous membrane tubing 4, MWCO: 12–14000) were acquired from Spectrum Laboratories, Inc. and were soaked in water for 30 minutes before use. Deuterated chloroform (99.8 atom% D, 0.5 wt% silver foil as stabilizer) was from Aldrich. Durapore PVDF membrane, 0.22 μm pore size filters were from Millex GV.
Synthesis
The 8-arm poly(ethylene glycol) end-group modified with terpyridyl groups was synthesized by the following procedure. Ground potassium hydroxide (KOH, 0.68 g, 12.1 mmol) and 8PEG (5.90 g, 0.30 mmol) were dissolved in 240 mL of dimethyl sulfoxide (DMSO) at 60 °C in a N2 atmosphere. After 90 min 4′-chloro-2,2′:6′,2′′-terpyridine (0.5 g, 1.87 mmol) was added and the reaction mixture was stirred for another 90 min at 60 °C, during which the color of the solution changed from dark red to orange. The reaction mixture was cooled and the products precipitated in diethyl ether. After decantation, the sticky, slightly orange product was mixed with 125 mL of brine. The resulting mixture was extracted 4 times with 250 mL of chloroform and the combined organic layers were dried over sodium sulfate. After filtration, the chloroform was evaporated under reduced pressure and the remaining liquid was dissolved in dichloromethane and precipitated in cold diethyl ether. The product was dried in vacuum at room temperature (yield: 4.8 g; 77%). Additional purification of the product was performed by dissolving the polymer in water and subsequent dialysis against water for 24 hours. The 8PEG(tpy)5.4OH2.6 was recovered by lyophilization. 1H NMR (400 MHz, CDCl3): δ = 3.63 (PEG protons), 3.92 (t, 2H, OCH2CH2O-terpyridyl), 4.46 (t, 2H, OCH2CH2O-terpyridyl), 7.32 (m, 2H, terpyridyl), 7.84 (dd, 2H, terpyridyl), 8.03 (s, 2H, terpyridyl), 8.60 (d, 2H, terpyridyl), 8.69 (d, 2H, terpyridyl).Methods
700, 324), FeCl2 (0.50, 13400, 556), CoCl2 (0.25, 20600, 307), and ZnCl2 (0.20, 27800, 322).
:2 mol mol−1) in water were prepared at a concentration of 0.78 mg mL−1. Subsequently, the resulting solution was added to a fused quartz cuvette and a 100 times molar excess of NiCl2·6H2O or FeCl2 in 50 μL of water was added. The change in the absorption at 556 nm (exchange of Fe2+ by Ni2+) or increase in the absorption at 556 nm (exchange of Co2+ by Fe2+) were recorded over time at room temperature.
:8, 3:8, 1:2, 3:4 and 1:1. After addition of an aliquot of the metal ion solution the resulting solution was equilibrated overnight. Size distributions were recorded on a Malvern Instruments Zetasizer Nano ZS at 20 °C and a 173° backscatter angle. Solutions containing a M2+–terpyridyl molar ratio of 1:2 were used to measure the zeta potential. The zeta potentials were recorded (Malvern Instruments Zetasizer Nano ZS) at 20 °C, using a 633 nm He–Ne laser and backscattering detection. Polystyrene capillary folded cells were used.
:2). The gels were left overnight at room temperature. A gel was placed at the center of the measuring plate at 5 °C and the measuring gap was set to 0.3 mm, and excess gel was removed. The system was equipped with an oil based solvent trap and the storage (G′) and loss modulus (G′′) were recorded under oscillatory shear. Measurements were performed at a frequency (ω) of 1 Hz and strain (γ) of 1%. Subsequently, heating and cooling cycles were performed between 5 and 60 °C at a rate of 1 °C min−1. After the temperature cycles, samples were cooled to 20 °C and a frequency and amplitude sweep were performed between ω = 0.1–100 Hz (γ = 1%) and γ = 0.01–100% (ω = 1 Hz) to confirm that all samples were within the linear viscoelastic regime (Fig. S4 and S5†).
Results and discussion
The hydroxyl groups of the star shaped 8PEG were partly converted into terpyridyl groups by a nucleophilic aromatic substitution reaction of potassium hydroxide generated alkoxide groups of the 8PEG to 4′-chloro-2,2′:6′,2′′-terpyridine as depicted in Fig. 1. The reaction was performed for 1 h and within this time period a color change can be observed from dark red to orange. The final ratio of hydroxyl to terpyridyl groups was controlled by the molar feed ratio of the reactants, although full conversion of the hydroxyl groups could not be obtained when an excess of the chloro-terpyridine was used. The reaction was performed in such a way that approximately 5 of the 8 hydroxyl groups were substituted with terpyridyl groups. The 1H-NMR spectrum confirmed the structure of the product (Fig. 2) and the number of substituted hydroxyl groups was calculated from the ratio of the terpyridine protons (a–e) and the CH2-groups of the terminal PEG oxy ethyl unit (f and g). Of the 8 hydroxyl groups initially present on average 5.4 hydroxyl groups per polymer molecule were substituted by terpyridyl groups affording 8PEG(tpy5.4)OH2.6. | ||
| Fig. 1 Synthesis of an 8PEG partially substituted with terpyridyl groups. | ||
 | ||
| Fig. 2 1H-NMR spectrum of 8PEG(tpy5.4)OH2.6 (CDCl3). | ||
To determine complexation ratios of the 8PEG(tpy5.4)OH2.6 with Ni(II), Fe(II), Co(II) and Zn(II) ions in water, UV-Vis titration experiments were carried out. Moreover, given the known high kinetic stability of the Ni2+ bis(terpyridine) complexes, the titration with a NiCl2 solution was used to determine the average number of terpyridyl-groups per polymer molecule. Upon titration, the evolution of the characteristic adsorption band of the Ni2+-complex at 324 nm (Fig. S1A†) was used to determine the molar complex ratio. By definition, the Ni2+-complex absorption will reach a maximum when all terpyridyl groups are complexed. As depicted in Fig. 3A, at a ratio of Ni2+ to terpyridyl-groups of 1:2, the absorption indeed remains constant upon further addition of Ni2+ ions. The number of terpyridyl groups coupled to the 8PEG can then be calculated from this maximum (i.e., from the intercept of the straight lines). The average number of coupled terpyridyl groups was calculated as 5.4, which is similar to that which was determined by 1H-NMR.
 | ||
| Fig. 3 UV-Vis absorption spectra recorded for the titration of a 8PEG(tpy5.4)OH2.6 solution with a NiCl2 (A) and CoCl2 solution (B). Absorption values were determined at 324 nm (Ni2+) and at 307 nm (Co2+). | ||
Titrations with the other transition metal chlorides, Fe2+, Co2+, and Zn2+ were performed similarly. From the characteristic Fe2+, Co2+ and Zn2+ bis-terpyridyl metal ligand charge transfer absorption bands at 556 nm, 307 nm and 322 nm, respectively, the complex ratios were determined (Fig. 3B and S1, S2†). Titration with Fe2+ also demonstrated a maximum complexation ratio of 1:2, whereas the metal ion–terpyridyl-groups ratios were somewhat higher for the Co2+ and Zn2+ complexes. This is a clear indication that Fe2+ forms stable bis-terpyridyl complexes with 8PEG(tpy5.4)OH2.6 in water, whereas in the case of Co2+ or Zn2+ mono-complexes may also be present.34 Moreover, the decrease of the absorption at higher Co2+–terpyridyl ratios is indicative of a lower stability of cobalt complexes compared to iron and nickel complexes. Similarly, adding an excess of Zn2+ ions results in a decrease of the absorption at 322 nm, indicative of the formation of mono-complexes.20
In the titration experiments, complex formation appears quickly and may even be diffusion controlled. Rate constants for complex formation of transition metal ions and terpyridyl-groups have been determined in the past and are generally 104 M−1 s−1.22 The differences observed in the titration experiments with an excess of metal ions are therefore indicative of differences in the dissociation rate constants. Because the dissociation rate constants were expected to have an influence on the aggregation at dilute conditions and crosslinking into hydrogels at higher concentrations (vide infra), the dissociation rate constants of the transition metal complexes of the polymers in water were first determined.
Metal exchange experiments in dilute solutions were performed according to a procedure described by Henderson and Hayward.33 Fe2+–bis(terpyridyl) complexes were dissociated with a 100-fold excess Ni2+ and of Co2+–bis(terpyridyl) complexes were dissociated with a 100-fold excess Fe2+. In both cases, the absorption band of the Fe2+–bis(terpyridyl) complexes was followed by UV-Vis spectroscopy. Henderson and Hayward have shown that these exchanges enable the determination of dissociation rate constants of the Fe2+ and Co2+ bis(terpyridyl) complexes.
The dissociation and association of the metal ion bis(terpyridyl) complexes in the presence of an excess of stronger complexing metal ions can be described by a series of equilibrium reactions as depicted in Fig. 4. During metal ion exchange the initial bis-complex dissociates (equilibrium 1) and the resulting free terpyridyl group rapidly forms a mono-complex with the stronger complexing metal ion (equilibrium 2). The mono-complexes subsequently disproportionate and bis-complexes are finally formed (equilibrium 3).
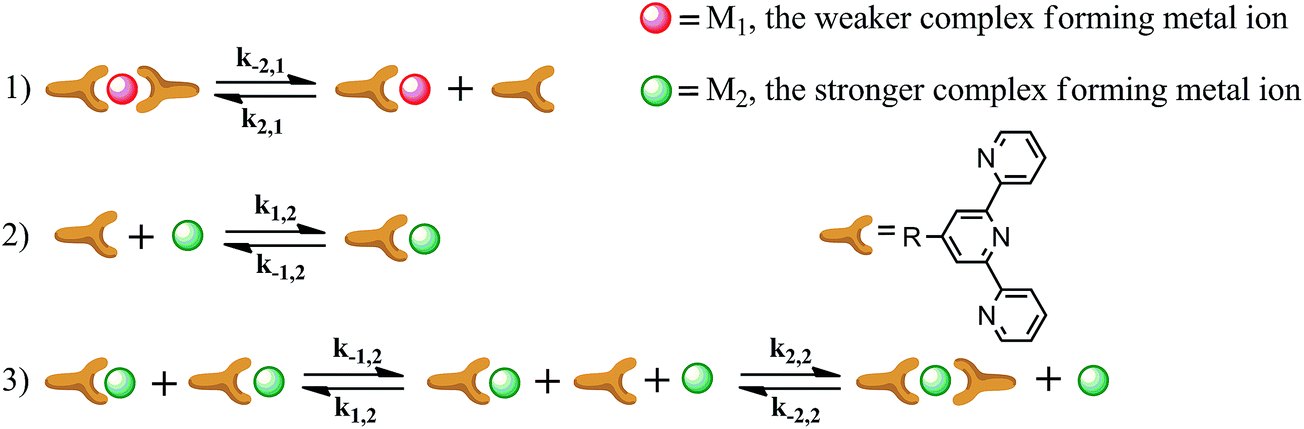 | ||
| Fig. 4 Equilibria involved in the metal ion exchange of bis(terpyridyl) complexes. Rate constants are indicated by km,n, in which m refers to a mono (1) or bis-complex (2) and n refers to the metal ion as depicted in the figure. | ||
Equations describing the changes in Fe2+–bis(terpyridyl) concentration in time were derived for either the exchange of Fe2+–bis(terpyridyl) to Ni2+–bis(terpyridyl) complexes (eqn (1)) or Co2+–bis(terpyridyl) to Fe2+–bis(terpyridyl) complexes (eqn (2)). Details for deriving these equations are given in S3.† In these equations, k−2 is the dissociation rate constant in s−1 and t is time in sec. Using Matlab® scripts, eqn (1) and (2) were fitted to the collected data (Fig. 5).
| [Fe2+(tpy)2]t = [Fe2+(tpy)2]0e−k−2,Fet | (1) |
| [Fe2+(tpy)2]t = 1/2[Co2+(tpy)2]0(1 − e−k−2,Cot) | (2) |
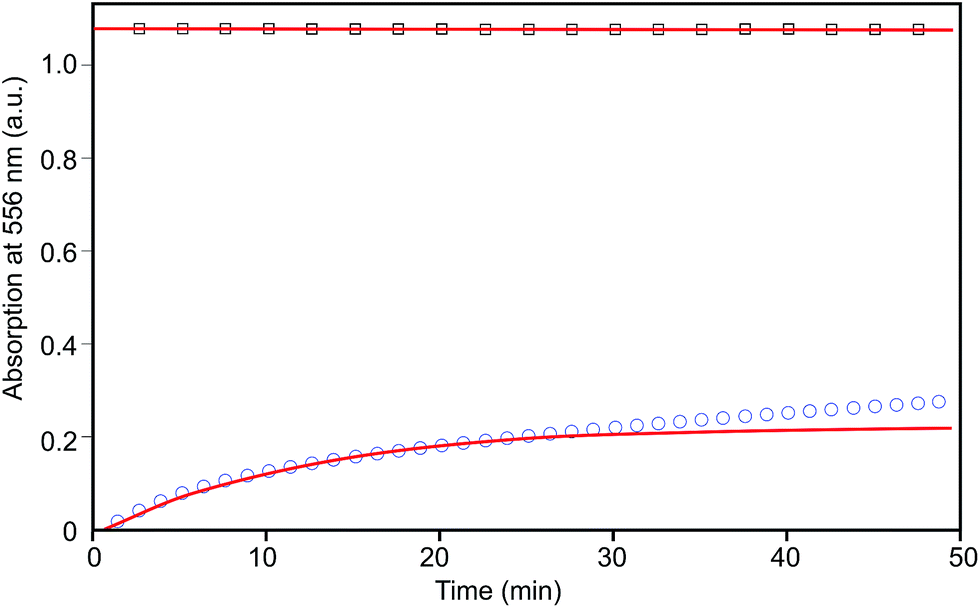 | ||
| Fig. 5 Absorption at 556 nm as a function of time of the Fe2+ to Ni2+ exchange (squares) and of the Co2+ to Fe2+ exchange (circles). Lines represent fits of the measured data. | ||
The exchange of Co2+ by Fe2+ ions from the Co2+–bis(terpyridyl) complex proceeds relatively quickly and could be accurately fit over the first 20 minutes of the metal ion exchange. Within this time span, metal ion exchange is dominated by Co2+–bis(terpyridyl) dissociation. Thereafter, the data deviate from the fit due to Co2+–mono(terpyridyl) dissociation. The fit yields a dissociation rate constant k−2,Co of 1.4 × 10−3 s−1, similar to the value presented in literature for the dissociation rate constant of a Co2+ complex of a linear terpyridyl end-group modified PEG (1.5 × 10−3 s−1).33 The exchange of Fe2+ by Ni2+ ions from the Fe2+–bis(terpyridyl) complex proceeds rather slowly and the data could be fit according to eqn (1) over at least 6 hours, yielding a dissociation rate constant of 8.1 × 10−7 s−1. This dissociation rate constant is lower than that determined for an Fe2+ complex of terpyridyl end-group modified linear PEG (5.0 × 10−6 s−1) but higher than that of an unsubstituted terpyridine (1.6 × 10−7).22,33 The differences observed in the dissociation rate constants could be a result of more effective shielding of the bis(terpyridyl) complexes by the 8-arm polymer. This may limit the accessibility of the complexes by metal ions in solution. The dissociation rate constants of bis(terpyridyl) complexes with Ni2+ and Zn2+ could not be determined. For the Ni2+ complexes this is due to their high stability. The Zn2+ complexes have too low a stability and mainly free ligands are present (equilibrium 1 in Fig. 4 is to the right).
In dilute aqueous solutions (3 mg mL−1), 8PEG-TEP5.4OH2.6 forms nano-particles with an average diameter of ∼10 nm. Please note that intensity plots do not provide ratios of particle size distributions, as larger particles scatter more than small particles. However, such plots give insight into changes in aggregation phenomena that can be caused by structural changes. The particle size of the 8PEG-TEP5.4OH2.8 as measured with DLS appeared similar to that of the 8 armed PEG(OH)8 revealing that the polymer does not aggregate in water at dilute conditions. Titration of 8PEG(tpy5.4)OH2.6 with the transition metal ions Ni2+ and Fe2+ resulted in a distinct shift of the average particle size (Fig. 6). Upon titration, a new distribution of particles with an average size of ∼200 nm appears, whereas the distribution of the small particles of ∼10 nm shifts to lower values with decreased intensities. A high resolution SEM picture is presented in the ESI (Fig. S6).†
 | ||
| Fig. 6 Nano-particle formation of 8PEG(tpy5.4)OH2.6 with Ni2+ (top) or Fe2+ (bottom) chlorides at different ratios probed by DLS. | ||
The distinct change in size distribution indicates the formation of Ni2+–terpyridyl complexes disrupting the hydrophobic interactions between terpyridyl groups and causing rearrangement to minimize the surface free energy. At a Ni2+–terpyridyl-groups ratio of 1:2 groups the smaller particles are no longer present and addition of an excess metal ions does not result in changes of the particle size distribution. The decrease in the average diameter of the small particles (∼10 nm) upon addition of Ni2+ ions is similar as observed by Chiper et al. on terpyridyl end-group modified pluronics.27 They explained this decreasing average particle size by rearrangement into intramicellar complexes, generating flower type micelles. Here, the distinct change to particles with an average diameter of ∼200 nm upon titration also reveals a fast rearrangement. Likely the Ni2+–terpyridyl complexes formed rearrange to the outer surface of the particles. Both the charge distribution and shielding of charges by PEG may contribute to the stability of the particles (vide supra).
Contrary to the distinct complete change in size distribution upon titration with Fe2+ or Ni2+ chlorides, the titration with Co2+ or Zn2+ chlorides yields nano-particle suspensions with multiple size distributions (Fig. 7). Similarly as described above, upon the addition of Co2+ the average size of the small particles is shifted to lower values. However, the formation of larger particles is less pronounced. Intermediate sized particles (average diameter of 20–100 nm) are observed during titration at Co2+–terpyridyl-groups ratios of 1:4 and 3:8. Furthermore, small micellar type nano-particles with an average diameter of 5 nm are still present at a Co2+–terpyridyl ratio of 1:2. Even adding an excess of Co2+ relative to the amount of terpyridyl-groups results in a narrowing of the particle size distribution at 200 nm diameter while the small particle distribution at 5 nm still present (data not shown). The larger variation of the nano-particle size distribution is likely caused by the relatively low stability of the Co2+–terpyridyl complexes compared to the Ni2+–terpyridyl and Fe2+–terpyridyl complexes in water. Possibly, exchange of Co2+ ions between terpyridyl groups occurs, allowing the formation of both mono-complexes and bis-complexes into intermediate sized particles. Upon addition of distinct volumes of a Zn2+ chloride solution, similar changes were observed. The multimodal size distributions observed indicate that both mono and bis-complexes are present at equilibrium. At higher metal ion to terpyridyl-group ratios only small changes were observed in the size distribution of the Zn2+ and Co2+ complexes, the distribution at 200 nm becoming more narrow, whereas the distribution of the Fe2+ and Ni2+ complexes did not change.
 | ||
| Fig. 7 Nano-particle formation of 8PEG(tpy5.4)OH2.6 with Co2+ (top) and Zn2+ (bottom) chlorides at different ratios probed by DLS. | ||
The changes in size distribution by complex formation of the terpyridyl-groups with transition metal ions indicate the reorganization of the initially present micelles. The zeta potentials were determined at a metal ion–terpyridyl ratio of 1:2 and revealed significant differences between the particle or particle distributions formed with Fe2+ or Ni2+ and those formed with Zn2+ or Co2+. For the latter the determined zeta potentials gave an average positive surface charge of 5 and 8 mV, respectively. The Ni2+ and Fe2+–terpyridyl complexes form only larger particles at this ratio providing a larger positive charge of 17–18 mV. Although particle suspensions prepared by Ni2+ and Fe2+–terpyridine complexation are more stable than those comprising Zn2+ or Co2+ complexes, they will still tend to flocculate over time as their zeta potential is lower than 30 mV.
Addition of transition metal ions to aqueous solutions of 8PEG(tpy5.4)OH2.6 at concentrations higher than 3 wt% produced hydrogels instead of discrete particles. Gelation took place instantaneously. Previous research on three and four armed poly(ethylene glycol)s having terpyridyl end-groups indicated that stable gels could be obtained with either Co3+ or Fe2+.26,30 but the mechanical properties of the gels were not studied. Considering the large differences in aggregation behavior, we studied the temperature dependent mechanical properties of the hydrogels by oscillatory rheology (see Table 1). The effect of temperature on the gels was assessed by measuring changes in the storage (G′) and loss (G′′) moduli between 5 and 60 °C applying cyclic temperature measurements. Such cyclic measurements can show that the gels are in equilibrium and may show temperature dependent gelation. The mechanical properties presented in Table 1 are from the last cooling cycle.
| Metal ion | Polymer (wt%) | Temperature (°C) | G′ (Pa) | G′′ (Pa) | tanδ × 103 |
|---|---|---|---|---|---|
| Ni2+ | 5 | 10 | 3100 | 4 | 1.3 |
| 60 | 6100 | 8 | 1.3 | ||
| 10 | 10 | 9300 | 13 | 1.4 | |
| 60 | 8200 | 50 | 6.1 | ||
| Fe2+ | 5 | 10 | 3200 | 10 | 3.1 |
| 60 | 5500 | 20 | 3.7 | ||
| 10 | 10 | 8200 | 18 | 2.2 | |
| 60 | 12500 |
35 | 2.8 | ||
| 20 | 10 | 21000 |
56 | 2.7 | |
| 60 | 28000 |
116 | 4.1 | ||
| Co2+ | 5 | 10 | 3600 | 17 | 4.7 |
| 60 | 2700 | 230 | 85 | ||
| 10 | 10 | 5100 | 8 | 1.5 | |
| 60 | 6100 | 250 | 41 | ||
| 20 | 10 | 6900 | 10 | 1.1 | |
| 60 | 8900 | 340 | 38 | ||
| Zn2+ | 5 | 10 | 2000 | 620 | 310 |
| 60 | 7 | 55 | 7900 | ||
| 10 | 10 | 13000 |
3100 | 240 | |
| 60 | 110 | 1200 | 11000 |
||
| 20 | 10 | 28000 |
6900 | 250 | |
| 60 | 8800 | 5000 | 570 |
The damping factor of the Ni2+ and Fe2+ gels varies from approximately 1 × 10−3 to 5 × 10−3, which demonstrates that elastic hydrogels were obtained. The much higher damping factor of the Co2+ gels at higher temperatures and Zn2+ gels reveals a much more viscous behavior.
As depicted in Fig. 8 the storage moduli of the Ni2+ and Fe2+ gels increase with an increase in temperature according the theory of rubber elasticity (eqn (3)). In this equation G′ is the storage modulus in Pa, ρ is the cross-link density in moles of elastically effective network chains per m3, R is the gas constant (8.314 J K−1 mol−1) and T is the temperature in K.
| G′ = ρRT | (3) |
 | ||
Fig. 8 Temperature dependent storage (G′, ![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and loss (G′′, ) and loss (G′′, ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) modulus of Ni2+ and Fe2+ 8PEG(tpy5.4)OH2.6 (M2+–terpyridyl = 1:2) hydrogels upon heating to 60 °C and subsequent cooling to 5 °C (three cycles) at different concentrations. Blue, 20 wt%, red, 10 wt% and black, 5 wt%. ) modulus of Ni2+ and Fe2+ 8PEG(tpy5.4)OH2.6 (M2+–terpyridyl = 1:2) hydrogels upon heating to 60 °C and subsequent cooling to 5 °C (three cycles) at different concentrations. Blue, 20 wt%, red, 10 wt% and black, 5 wt%. | ||
Depending on concentration, the Co2+ gels show a different change in storage and loss modulus with temperature. The damping factors of the Co2+ gels are approximately twenty five-fold higher at 60 °C than at 10 °C. As illustrated in Fig. 9, the storage modulus of the 10 wt% gel increases with temperature similarly as observed for the Ni2+ and Fe2+ gels. Contrary, 5 wt% Co2+ gels show an initial increase in the storage modulus between 5 and 25 °C followed by a decrease upon further heating. This is a clear indication that temperature affects the number of effective crosslinks in the gel.
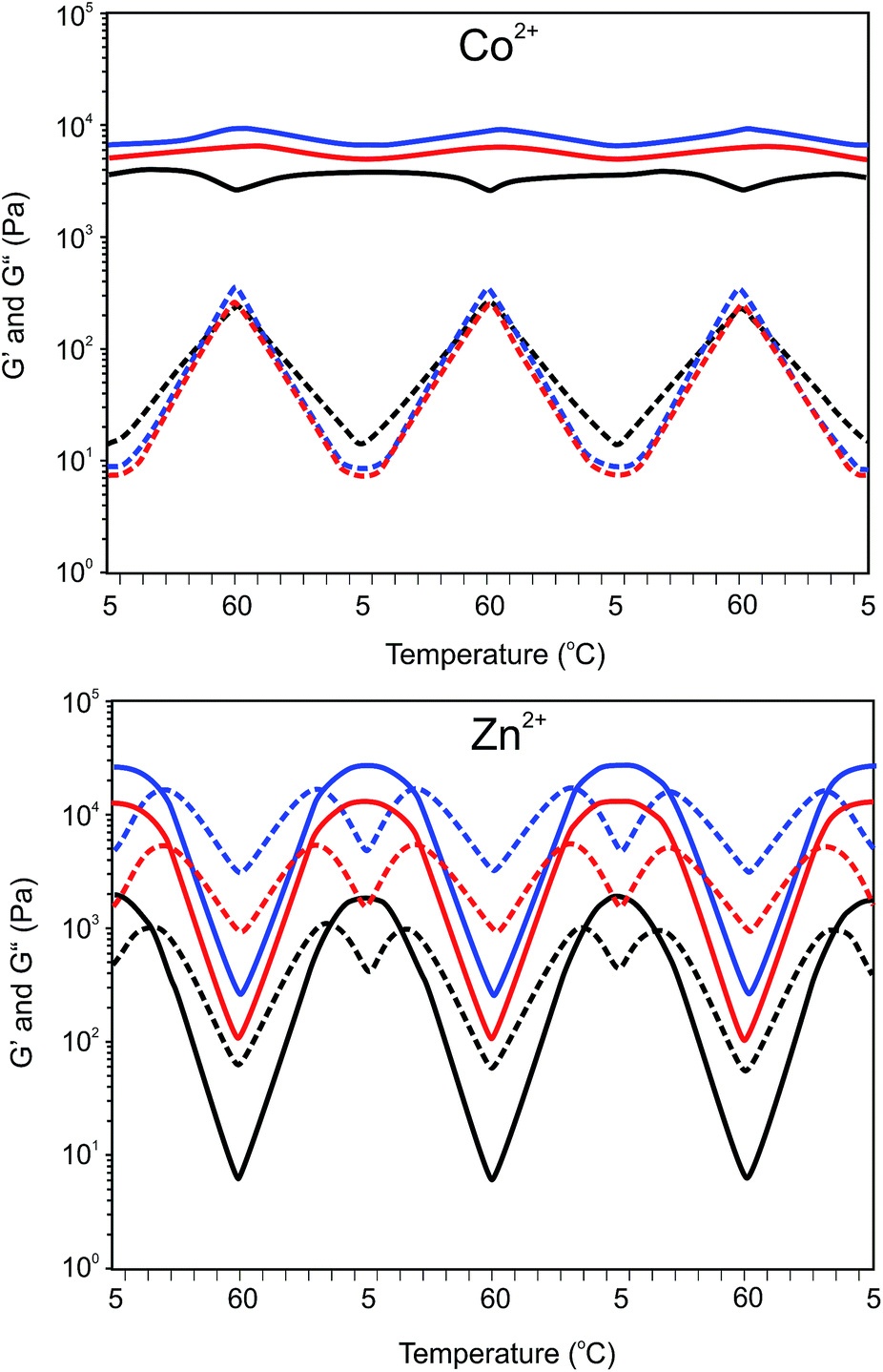 | ||
| Fig. 9 Temperature dependent storage (G′, ) and loss (G′′, ) modulus of a Co2+ or Zn2+ 8PEG(tpy5.4)OH2.6 (M2+–terpyridyl = 1:2) hydrogel upon heating to 60 °C and subsequent cooling to 5 °C (three cycles). Blue, 20 wt%, red, 10 wt% and black, 5 wt%. | ||
This becomes even more pronounced for the Zn2+ gels. Increasing the temperature results in a gel to sol transition. The transition from a gel to sol occurs at approximately at 25 °C and is reversible upon cooling. Within the temperature range investigated, the storage modulus decreases with increasing temperatures, whereas the loss modulus increases up to the transition temperature and decreases upon further heating. This trend is witnessed for all Zn2+ gels at different concentrations. These results suggest that the increasing elastic modulus upon a temperature increase is counteracted by a decrease in elastic contribution due to dissociation of complexes. Thus, although temperature increases the modulus, the equilibrium cross-link density decreases, resulting in an overall decreasing modulus. Moreover, the high modulus of the Zn-complexes is likely the result of efficient crosslinking. This can be due to the lability of Zn-complexes formed, which allows easy reorganization. This is also reflected in the rather high loss modulus and thus less elastic networks formed. At higher concentrations the mechanical properties of the Zn gels are increasing due to a higher crosslink density.
Interestingly, the gel to sol transition temperature of the Zn2+ gels does not change significantly with polymer content even though the modulus does increase at higher concentrations. The increasing cross-link density with polymer content resulting in an increased modulus is in accordance with the rubber elasticity theory. The temperature independent and reversible sol to gel transition suggests that more cross-links per unit volume are disrupted upon increasing temperatures in the higher polymer content gels compared to gels with a lower polymer content. At this moment we only can give a hypothesis to this phenomenon. The mechanical stability of hydrogels depends on the number of crosslinks formed and the inherent stability of the supramolecular crosslinks. Assuming the association rate of complex formation is very high, hydrogel properties likely depend on the dissociation rate constant of the complexes which has an Arrhenius type temperature dependency. This is reasonable as the formation of terpyridine complexes is an equilibrium process. Thus, at a certain temperature, the ratio between mono-complex and bis-complex (i.e. cross-links) per unit volume will be independent of the polymer content and free polymer chain ends will be formed more rapidly in respect to temperature in the higher polymer content gels compared to those with a lower polymer content. Therefore, polymer chain mobility will increase more rapidly with temperature, yielding a similar transition temperature for a gel with higher polymer content compared to a gel with lower polymer content.
Conclusions
The differences in dissociation rate constants of a supramolecularly crosslinked terpyridine end-group modified 8-arm PEG (8PEG-terpyridyl) with transition metal ions is reflected in nanoparticle formation at low concentrations and hydrogel properties at concentrations above 5 wt%. Whereas Ni2+ or Fe2+ complexes at low concentrations afforded nanoparticles, Co2+ or Zn2+ complexes were mainly present as micellar type aggregates. Hydrogels formed by 8PEG-terpyridyl with Ni2+ and Fe2+ were elastic and showed minor changes with temperature between 5 and 60 °C. Due to the low kinetic stability of the Co2+ and Zn2+ complexes, the loss modulus of the hydrogels is largely influenced by temperature and led for the Zn2+ complexes to a reversible sol gel transition at 25 °C. Importantly, the sol–gel transition temperature of the Zn2+ complexes was independent of polymer content.References
- J. Aleman, A. V. Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvil, I. Meisel, I. Mita, G. Moad, S. Penczek and R. F. T. Stepto, Pure Appl. Chem., 2007, 79, 1801–1827 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd., 2nd edn, 2009, pp. 27–36.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- E. J. Dobratz, S. W. Kim, A. Voglewede and S. S. Park, Arch. Facial Plast. Surg., 2009, 11, 40–47 CrossRef PubMed.
- Y. J. Tang, J. Sun, H. S. Fan and X. D. Zhang, Carbohydr. Polym., 2012, 88, 46–53 CrossRef CAS PubMed.
- J. K. F. Suh and H. W. T. Matthew, Biomaterials, 2000, 21, 2589–2598 CrossRef CAS.
- A. J. Goshe, J. D. Crowley and B. Bosnich, Helv. Chim. Acta, 2001, 84, 2971–2985 CrossRef CAS.
- J. Brassinne, C. A. Fustin and J. F. Gohy, J. Inorg. Organomet. Polym. Mater., 2013, 23, 24–40 CrossRef CAS.
- T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS PubMed.
- W. G. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS PubMed.
- C. J. Jones, Supramolecular Chemistry, Royal Society of Chemistry, Cambridge, 1st edn, 2002, ch. 4, pp. 54–70.
- M. Albrecht, Naturwissenschaften, 2007, 94, 951–966 CrossRef CAS PubMed.
- B. König, Vorlesung Supramolekulare Chemie, Regensburg, 2009, pp. 1–72 Search PubMed.
- E. C. Constable, Chem. Soc. Rev., 2007, 36, 246–253 RSC.
- A. C. Benniston, A. Harriman, D. J. Lawrie, M. Mehrabi and O. D. Russell, Inorg. Chim. Acta, 2005, 358, 3483–3490 CrossRef CAS PubMed.
- E. C. Constable, J. Healy and M. G. B. Drew, Polyhedron, 1991, 10, 1883–1887 CrossRef CAS.
- M. Satterfield and J. S. Brodbelt, Inorg. Chem., 2001, 40, 5393–5400 CrossRef CAS PubMed.
- L. Kosbar, C. Srinivasan, A. Afzali, T. Graham, M. Copel and L. Krusin-Elbaum, Langmuir, 2006, 22, 7631–7638 CrossRef CAS PubMed.
- R. Shunmugam, G. J. Gabriel, K. A. Aamer and G. N. Tew, Macromol. Rapid Commun., 2010, 31, 784–793 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, d- and f-Block Chemistry (Basic Concepts in Chemistry), John Wiley & Sons, Ltd., 2nd edn, 2009, pp. 1–48.
- R. H. Holyer, C. D. Hubbard, S. F. A. Kettle and R. G. Wilkins, Inorg. Chem., 1966, 5, 622–625 CrossRef CAS.
- I. M. Henderson and R. C. Hayward, J. Mater. Chem., 2012, 22, 21366–21369 RSC.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- B. G. G. Lohmeijer and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1413–1427 CrossRef CAS.
- S. Schmatloch and U. S. Schubert, Macromol. Symp., 2003, 199, 483–497 CrossRef CAS.
- M. Chiper, S. Hoeppener, U. S. Schubert, C. A. Fustin and J. F. Gohy, Macromol. Chem. Phys., 2010, 211, 2323–2330 CrossRef CAS.
- F. D. Jochum, J. Brassinne, C. A. Fustin and J. F. Gohy, Soft Matter, 2013, 9, 2314–2320 RSC.
- M. Kimura, Y. Nakagawa, N. Adachi, Y. Tatewaki, T. Fukawa and H. Shirai, Chem. Lett., 2009, 38, 382–383 CrossRef CAS.
- T.-A. Asoh, H. Yoshitake, Y. Takano and A. Kikuchi, Macromol. Chem. Phys., 2013, 214, 2534–2539 CrossRef CAS.
- T. Ueki, Y. Takasaki, K. Bundo, T. Ueno, T. Sakai, Y. Akagi and R. Yoshida, Soft Matter, 2014, 10, 1349–1355 RSC.
- T. Rossow and S. Seiffert, Polym. Chem., 2014, 5, 3018–3029 RSC.
- I. M. Henderson and R. C. Hayward, Polym. Chem., 2012, 3, 1221–1230 RSC.
- R. Hogg and R. G. Wilkins, J. Chem. Soc., 1965, 1, 341–350 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sm01162g |
| This journal is © The Royal Society of Chemistry 2014 |