 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lamellar and liquid crystal ordering in solvent-annealed all-conjugated block copolymers†
Yen-Hao
Lin
a,
Kevin G.
Yager
b,
Bridget
Stewart
a and
Rafael
Verduzco
*a
aDepartment of Chemical and Biomolecular Engineering, MS-362, Rice University, 6100 Main Street, Houston, Texas 77005, USA. E-mail: rafaelv@rice.edu
bCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA
First published on 31st March 2014
Abstract
All-conjugated block copolymers are an emerging class of polymeric materials promising for organic electronic applications, but further progress requires a better understanding of their microstructure including crystallinity and self-assembly through micro-phase segregation. Here, we demonstrate remarkable changes in the thin film structure of a model series of all-conjugated block copolymers with varying processing conditions. Under thermal annealing, poly(3-hexylthiophene)-b-poly(9′,9′-dioctylfluorene) (P3HT-b-PF) all-conjugated block copolymers exhibit crystalline features of P3HT or PF, depending on the block ratio, and poor π–π stacking. Under chloroform solvent annealing, the block copolymers exhibit lamellar ordering, as evidenced by multiple reflections in grazing incidence wide- and small-angle X-ray scattering (GIWAXS and GISAXS), including an in-plane reflection indicative of order along the π–π stacking direction for both P3HT and PF blocks. The lamellae have a characteristic domain size of 4.2 nm, and this domain size is found to be independent of block copolymer molecular weight and block ratio. This suggests that lamellar self-assembly arises due to a combination of polymer block segregation and π–π stacking of both P3HT and PF polymer blocks. Strategies for predicting the microstructure of all-conjugated block copolymers must take into account intermolecular π–π stacking and liquid crystalline interactions not typically found in flexible coil block copolymers.
Introduction
All-conjugated block copolymers are an emerging class of materials comprised of two or more covalently linked conjugated polymer chains. This class of block copolymers is of interest for organic electronic applications because they combine the optoelectronic properties of semiconductive polymers with structure control through micro-phase segregation1,2 and crystallization.3,4 As an example, recent work demonstrated significant performance enhancement in block copolymer organic photovoltaics (OPVs) compared with polymer–polymer blends.5 However, further progress in the development of all-conjugated block copolymers requires a better understanding of the microstructure of all-conjugated block copolymers and how their crystallinity, micro-phase segregation, and domain orientation can be controlled by applying different processing strategies.Comprehensive theoretical and experimental studies of rod–coil diblock copolymers (containing one conjugated polymer block and one flexible coil polymer block) have revealed an interplay between relative block size, polymer crystallinity, liquid crystal ordering, and micro-phase segregation.6–11 Processing conditions have been demonstrated to give some control over the microstructure of rod–coil block copolymers, including aligned domains through the application of a magnetic field12–15 or the formation of micellar assemblies using a selective solvent.16 This work has led to the development of a quantitative model to describe the phase behavior of rod–coil block copolymers, including self-assembly and liquid crystal ordering.17 Comparable predictive models are unavailable for all-conjugated block copolymers due in part to limited experimental studies on structure–processing relationships. Previous work with P3HT-based all-conjugated block copolymers have found that P3HT crystallization dominates the film morphology, suppressing micro-phase segregation and crystallization of the second polymer block.18,19 Evidence for micro-phase segregation in rod–rod all-conjugated block copolymer has been inconclusive, potentially due to polymer crystallization, slow dynamics, stiff polymer backbones, and/or low enthalpic driving force for micro-phase segregation.20 Improved control over the microstructure of all-conjugated block copolymers and a broader understanding of structure–processing–property relationships could benefit their development for and use in organic electronic devices and applications.
Here, we report remarkable changes in the thin film structure of a model series of all-conjugated block copolymers under different annealing conditions. Thermally annealed poly(3-hexylthiophene)-b-poly(9′,9′-dioctylfluorene) (P3HT-b-PF) (Scheme 1) all-conjugated block copolymers exhibit crystalline features characteristic of P3HT or PF, but poor order along the π–π stacking direction. The same block copolymers, under solvent annealing, self-assemble into a lamellar phase, as evidenced by multiple reflections in grazing incidence wide- and small-angle X-ray scattering (GIWAXS and GISAXS). The degree of face-to-face π–π stacking substantially increases in solvent-annealed compared with unannealed films. We present a detailed analysis of the structure of P3HT-b-PF block copolymer films and the self-assembled lamellar phase, including paracrystallinity measurement, spectroscopic properties, and phase behaviour with temperature. These results indicate that all-conjugated block copolymers have a rich, processing-dependent microstructure determined by a combination of π–π stacking, crystallization, and micro-phase segregation of the polymer blocks.
 | ||
| Scheme 1 Chemical structure of poly(3-hexylthiophene)-b-poly(9′,9′-dioctylfluorene) (P3HT-b-PF). | ||
Experimental section
Synthesis of P3HT-b-PF
P3HT-b-PF block copolymers were synthesized as reported previously.21 Characteristics of P3HT-b-PF block copolymers reported in this study are listed in Table 1. 1H NMR spectra are provided in the ESI Fig. S1.†| Polymers | P3HTaMw (PDI) | BCPaMw (PDI) | DP ratiosb (P3HT wt%) |
|---|---|---|---|
| a M w (kg mol−1) and PDI for P3HT and block copolymers determined by comparison to a set of monodisperse polystyrene standards. Head-to-tail regioregularity of P3HT is greater than 93% for all samples as determined from 1H NMR. b DP ratios and P3HT content were determined by 1H NMR via comparison of the integrated intensity of P3HT aromatic peak (6.9 ppm) and fluorene alkyl peaks (2.2 ppm). | |||
| P3HT36-b-PF100 | 6.1 (1.16) | 48.4 (1.86) | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 (13%) :100 (13%) |
| P3HT81-b-PF105 | 13.5 (1.32) | 60.6 (1.87) | 81:105 (25%) |
| P3HT84-b-PF80 | 14.1 (1.10) | 42.5 (1.58) | 84:80 (33%) |
| P3HT84-b-PF13 | 14.1 (1.10) | 22.8 (1.39) | 84:13 (74%) |
Sample preparation and processing
Sample films were prepared by drop casting from 0.05 wt% chloroform solutions onto silicon substrates and quartz substrates. Evaporation of chloroform under ambient conditions gives films with a thickness of approximately 350 nm as measured by X-ray reflectivity. Samples were solvent annealed in a closed chamber saturated with chloroform at ambient temperature and pressure for 5 days. Samples were dried under vacuum for at least 12 hours prior to measurement.Grazing incident small- and wide-angle X-ray scattering (GIWAXS and GISAXS)
Grazing incidence small- and wide-angle X-ray scattering measurements were carried out on the X9 beamline at the National Synchrotron Light Source, Brookhaven National Laboratory. The undulator beamline was operated at an energy of 14 keV; two-dimensional images were collected using CCD area detectors. The beam size was 100 μm (h) × 50 μm (v). Sample detector distance was 370 mm for the wide-angle detector and 3091 mm for the small-angle detector. Sample measurements were carried out under vacuum which was in the range of 2–3 × 10−6 bar, with a temperature-controlled sample stage interfaced with a Lakeshore 340 unit. Where indicated, measurements were performed on sector 8-ID-E at Advanced Photon Source, Argonne National Laboratory. Details for these measurements are provided in the ESI.†Differential scanning calorimetry (DSC)
Differential scanning calorimetry measurements were performed using a TA Instrument DSC Q10 with a ramp rate of 5 °C min−1 under N2. Samples were placed in hermetic pans from Thermal Support, Inc. Solvent annealed samples were solvent annealed as described above for one month and dried under vacuum before measurement. All samples were subjected to at least two heating and cooling cycles between 10 °C to 250 °C.Ultraviolet-visible absorbance spectroscopy (UV-VIS)
UV-VIS measurements were carried out with a Shimadzu UV-3101PC spectrophotometer with scan range of 200–900 nm. All films were cast on quartz substrates.Polarized optical microscopy
Optical microscopy images of polymer films were acquired using an Axioplan 2 imaging microscope in reflective mode. Films were the same as films for GIXS measurements. Images were processed using Axio Vision version 4.8.Results and discussions
P3HT-b-PF is an all-conjugated block copolymer comprised of two semi-crystalline conjugated polymer blocks (crystal melting temperatures 220 °C and 150 °C for P3HT and PF, respectively). Prior work with P3HT-b-PF thin films has found a morphology dominated primarily by crystallization of the P3HT or PF block but no evidence for liquid crystal ordering or micro-phase segregation.19,21–23 We hypothesized that long-term solvent annealing in a good solvent for both blocks would increase chain mobility, yielding improved crystallinity, new phases and/or micro-phase segregation. Similar strategies applied to flexible coil block copolymers can result in improved long-range ordering of block copolymer domains and, in some cases, new phases through micro-phase segregation.24As detailed by prior work, the microstructure of P3HT and PF homopolymers is influenced by processing. Regioregular P3HT organizes into lamellar crystalline domains with face-to-face π–π stacking between chains and lamellar stacking through the hexyl side-chains.25 In solution processed thin films, the preferred orientation of P3HT crystallites can be dictated to some extent by varying processing conditions.26 PF exhibits crystalline, liquid crystalline, or amorphous phases depending on its processing history.27–29
To investigate the role of processing and annealing conditions, the microstructure of a series of P3HT-b-PF films with varying P3HT contents after solvent and/or thermal annealing were analyzed by grazing-incidence X-ray scattering, microscopy, UV-VIS absorbance measurements, and differential scanning calorimetry (DSC). P3HT-b-PF all-conjugated block copolymers with varying block ratios were synthesized via a combination of Grignard metathesis and Suzuki–Miyaura polycondensation. The P3HT-b-PF block copolymers studied have varying compositions ranging from 13 wt% of P3HT to 74 wt% of P3HT, as shown in Table 1. Samples films were drop cast from chloroform and analyzed by grazing-incidence wide- and small-angle scattering (GIWAXS and GISAXS) without annealing (as-cast), after thermal annealing, and with long-time solvent annealing (5 days at room temperature, CHCl3).
Grazing incidence X-ray scattering (GIXS) analysis of P3HT-b-PF under varying processing conditions
Small- and wide-angle X-ray scattering (GISAXS and GIWAXS) measurements provide information on both the in-plane (qxy, direction parallel to substrate) and out-of-plane (qz, direction perpendicular to the substrate) structure of the films. Peaks or reflections in the spectra indicate periodicities in the film microstructure, which may arise due to crystallinity or self-assembly processes. More information on the application of GIWAXS and GISAXS to study block copolymer films is provided by Ree et al.30GIWAXS measurements of P3HT-b-PF films are shown in Fig. 1 and ESI S2 and S3.† As-cast P3HT-b-PF films are amorphous and exhibit a weak in-plane reflection at q ∼ 1.45 Å−1 (see ESI Fig. S2 and S3†). This in-plane reflection matches the π–π stacking reflection observed in PF homopolymer films,31 and we therefore conclude this reflection indicates some π–π stacking of the PF polymer block in as-cast films. P3HT84-b-PF13 additionally exhibits weak scattering peaks characteristic of P3HT crystallites: an in-plane π–π stacking (010) peak at q ∼ 1.65 Å−1 and out-of-plane (100), (200), and (300) peaks.4,25,32
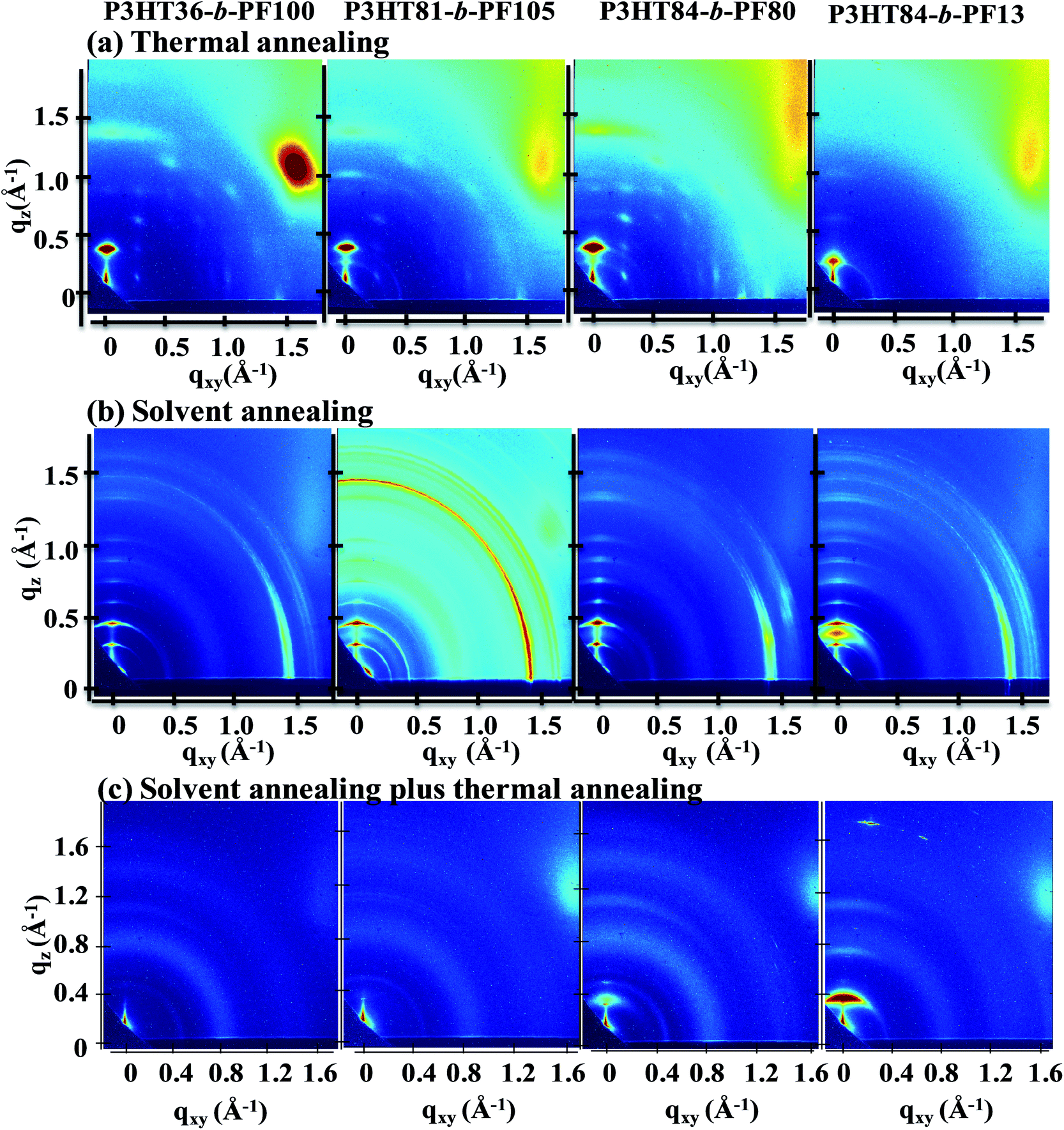 | ||
| Fig. 1 GIWAXS measurements for P3HT36-b-PF100, P3HT81-b-PF105, P3HT84-b-PF80, P3HT84-b-PF13 with varying processing histories, as indicated. (a) and (b) were acquired at room temperature and (c) was acquired at 150 °C. All samples were measured at an incident angle of 0.25° and 30 seconds exposure time. | ||
After thermal annealing at 220 °C – beyond the crystal melting temperatures for both blocks – and cooling to 100 °C, the films exhibit characteristics of pure P3HT or PF crystallites, or both, depending on the block ratio (Fig. 1a). Thermally annealed P3HT36-b-PF100 films exhibit characteristics of PF α-phase crystals33 while thermally annealed P3HT84-b-PF13 films show (100), (200), and (300) P3HT crystal reflections. Block copolymers with intermediate P3HT compositions, P3HT81-b-PF105 and P3HT84-b-PF80, show primarily reflections consistent with PF α-phase and some crystallization of the P3HT block. In contrast to thermally annealed P3HT homopolymer films, reflections corresponding to π–π stacking (q ∼ 1.45 Å−1 and 1.65 Å−1 for PF and P3HT, respectively) are absent for all thermally annealed block copolymer films.
Block copolymer films subjected to 5 days of solvent annealing in a good solvent for both blocks (chloroform) exhibit multiple out-of-plane reflections indicative of long-range, lamellar ordering (Fig. 1b, 2 and 3). A primary GISAXS reflection is observed at ∼0.15 Å−1, which corresponds to a spacing between lamellae of approximately 4.2 nm (Fig. 2). As shown in Fig. 3, the observed peaks are separated by integral multiples of q. These reflections reveal long-range lamellar ordering in solvent annealed P3HT-b-PF films not present in solvent or thermally annealed pristine P3HT or PF films. By contrast, solvent annealed P3HT homopolymer exhibits a (100) crystal peak while solvent-annealed PF films exhibit a metastable liquid crystal β mesophase34 (see ESI Fig. S4, S5, and S6†).
 | ||
| Fig. 2 GISAXS measurements for solvent-annealed P3HT36-b-PF100, P3HT81-b-PF105, P3HT84-b-PF80, P3HT84-b-PF13. Samples were analyzed at room temperature. | ||
 | ||
| Fig. 3 Linecuts of GISAXS (imbedded figure) and GIWAXS measurements along qxy ∼ 0 Å−1 for solvent annealed samples. | ||
Solvent-annealed block copolymer films also display strong in-plane reflections at q ∼ 1.45 Å−1 and q ∼ 1.65 Å−1 (see ESI Fig. S7†). These reflections are characteristic of face-to-face π–π stacking for PF and P3HT, respectively. Other crystal reflections characteristic of PF or P3HT homopolymers are absent in solvent annealed films, except for P3HT84-b-PF13. A peak corresponding to the (100) P3HT crystal spacing (q ∼ 0.39 Å−1) is clearly evident in solvent-annealed P3HT84-b-PF13 (see linecut in Fig. 3). A quantitative analysis of crystalline disorder through measurements of a paracrystallinity parameter is provided below.
On heating the solvent-annealed films from room temperature to 150 °C, lamellar GISAXS and GIWAXS reflections disappear and P3HT-b-PF films, with the exception of P3HT84-b-PF13, show only diffuse scattering, with no sharp reflections or peaks. This is indicative of an amorphous or liquid crystal nematic phase (Fig. 1c). Note that 150 °C is below the crystal melting temperatures for both P3HT and PF blocks.
The presence of nematic liquid crystal ordering is confirmed by inspection of the films by polarized optical microscopy (POM), which reveals a birefringent Schlieren texture characteristic of a nematic phase for P3HT36-b-PF100, P3HT81-b-PF105, and P3HT84-b-PF80 (Fig. 4a–c). The block copolymer with the highest P3HT content, P3HT84-b-PF13, (comprised of 85 wt% P3HT) does not exhibit a nematic phase. Instead, as shown in Fig. 1c, some P3HT crystallization is evident. POM analysis of all solvent-annealed and solvent plus thermal-annealed block copolymer films are provided in the ESI Fig. S8.†
 | ||
| Fig. 4 Polarized optical microscopy images of P3HT-b-PF block copolymer thin films subjected to solvent-annealing followed by thermal annealing at 150 °C. Images were acquired at room temperature. | ||
P3HT-b-PF films, with the exception of P3HT84-b-PF13, thus form a nematic phase after solvent annealing followed by heating to 150 °C and slowly cooling to room temperature. PF homopolymers exhibit a nematic phase only at temperatures higher than the crystal melting temperature35 or at room temperature after rapid quenching.28,36 The enhanced stability of the nematic phase in block copolymers may be due to poor chain mobility resulting from the rigidity of polymer chains or close π–π stacking in the self-assembled lamellar phase.37 The results also suggest that the lamellae of P3HT-b-PF is metastable and can be affected by elevated temperature as well as the crystallization of P3HT as shown in P3HT84-b-PF13.
P3HT-b-PF films thus exhibit remarkable changes depending on the processing history. Thermally annealed films show characteristics of P3HT or PF crystallites, but with poor order along the π–π stacking direction. Solvent-annealed films exhibit a self-assembled lamellar phase along with ordering along the π–π stacking direction. Solvent-annealed films subsequently heated to 150 °C transition to a nematic liquid crystal phase, which is stable on cooling back down to room temperature. This phase behaviour is distinct from that of P3HT or PF homopolymers. Below, we provide a proposed structure of the lamellar phase, detail the optical and thermal properties of the materials, and quantify disorder in solvent-annealed films through quantitative analysis of peak spacings and widths.
Schematic for lamellar assembly in solvent-annealed films
A schematic for the film microstructure consistent with the features of GISAXS and GIWAXS is shown in Fig. 5. P3HT and PF lamellar domains are oriented parallel to the substrate, and the primary lamellar peak reflection (qz ∼ 0.15 Å−1) (Fig. 2 and inset Fig. 3) corresponds to a domain spacing of 4.2 nm. Peaks at q ∼ 1.45 Å−1 and q ∼ 1.65 Å−1 correspond to face-to-face (π–π) packing for PF and P3HT, respectively, and indicate π–π stacking along the in-plane direction, as shown schematically in Fig. 5. The orientation of the face-to-face π–π stacking is perpendicular to the lamellar domains but free to rotate 180 degrees within the plane parallel to the substrate. For clarity, only one possible orientation is shown in the schematic.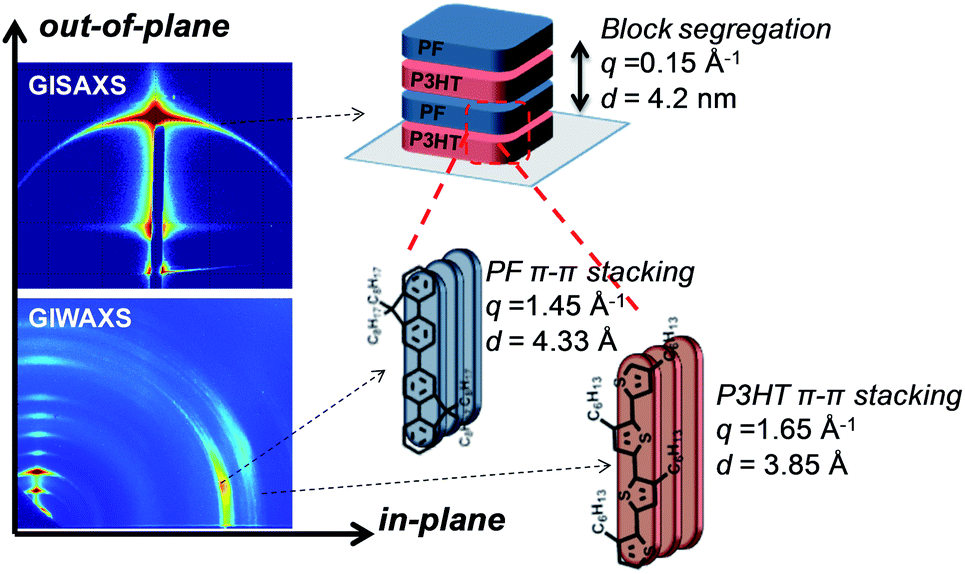 | ||
| Fig. 5 Representative GISAXS and GIWAXS reflections and schematic for lamellar ordering in solvent-annealed P3HT-b-PF all-conjugated block copolymers. | ||
The size of the lamellar domains formed after solvent annealing is invariant with block copolymer molecular weight and composition. All solvent-annealed block copolymers exhibit lamellar ordering with a characteristic domain spacing of 4.2 nm. The underlying mechanism for the formation of lamellar domains cannot be attributed to conventional micro-phase segregation as in coil–coil block copolymers, in which the domain spacing is dependent on the block copolymer molecular weight.38 Instead, we propose that self-assembled lamellae arise due to a combination of polymer block segregation and π–π interactions of both P3HT and PF polymer blocks. The lamellae are expected to consist of both ordered and amorphous regions, consistent with the structure of P3HT and other semicrystalline, conjugated polymers.39 Prior work has shown that P3HT polymer chains can be folded in amorphous regions and between crystalline domains, even with very sharp turns and folds of the P3HT backbone.40 This allows for a single chain to occupy both ordered and amorphous domains and pass in and out of relatively small domains. As a result, the lamellar domain size (4.2 nm) is determined primarily by π–π stacking associations and is relatively unaffected by block copolymer molecular weight and composition.
An alternative possibility for the structure of solvent-annealed block copolymer films is shown schematically in the ESI Fig. S9.† In this proposed schematic, polymer backbones are oriented in-plane rather than out-of-plane. As a result, the domain spacing is determined by the fully extended length of the alkyl side-chains. While this structure is consistent with the features observed by GISAXS and GIWAXS, we believe this type of ordering is unlikely due to the relatively large spacing between polymers, 4.2 nm. This would require stretching of the P3HT and PF alkyl side-chains. Additionally, the hypothetical ordering is not volume conserving due to the large space (4.2 nm) between polymer chains and close π–π stacking. As a result, the structure in Fig. 5 is the more likely possibility, but we acknowledge the potential for an alternative configuration shown in ESI Fig. S9.†
Optical and thermal properties of P3HT-b-PF films
UV-VIS absorbance analysis of block copolymer films can provide information about ordering and crystallinity, particularly along the π–π stacking direction. UV-VIS analysis has previously been used to study conformational changes in PF34,36 and P3HT.41–43 High-temperature thermally annealed block copolymer films, which exhibit a morphology dominated by crystallinity of either P3HT or PF (Fig. 1a), show only broad absorbance peaks at λ ∼ 390 and 500 nm, indicative of poor π–π stacking (Fig. 6a). Similar broad peaks by UV-VIS are observed in the PF crystalline α-phase34 and nematic phases.34 | ||
| Fig. 6 UV-VIS spectra for P3HT-b-PF block copolymers after: (a) high-temperature thermal annealing, (b) long-term solvent annealing, and (c) long-term solvent annealing followed by 150 °C thermal annealing. All spectra were acquired at room temperature. | ||
After solvent annealing, UV-VIS measurements reveal peaks at λ ∼ 522, 560 and 603 nm in the absorption spectra, reflecting P3HT π–π stacking in all solvent annealed films. Regioregular P3HT homopolymer, ordered in the π–π stacking direction, exhibits similar peaks by UV-VIS analysis.41–43 UV-VIS analysis also reveals peaks at λ ∼ 400 and 435 nm in the absorption spectra for lamellar block copolymer films, similar to what is seen for the PF β phase.31 This suggests that face-to-face π–π of PF and P3HT polymer blocks is enhanced in solvent-annealed films, compared with thermally annealed films.
After heating to 150 °C resulting in nematic liquid crystal ordering, sharp absorbance features characteristic of π–π stacking disappear. Nematic block copolymer films show only broad peaks in the absorption spectra at λ ∼ 390 and 500 nm, similar to thermally annealed block copolymer films and corresponding to poor face-to-face ordering of PF and P3HT blocks. The observed transformations in the UV-VIS spectra are thus consistent with GIWAXS measurements that indicate poor π–π stacking in crystalline and nematic block copolymer films and enhanced π–π stacking in the lamellar block copolymer phase.
Differential scanning calorimetry (DSC) reveals differences in the crystal melting temperatures for samples with different processing histories. With the exception of P3HT84-b-PF13, DSC measurements detect a phase transition at 145 °C in solvent-annealed block copolymer on the first heating cycle (Fig. 7a). This transition temperature is consistent with the transition to the nematic liquid crystal phase observed by GIWAXS.
 | ||
| Fig. 7 DSC analysis of P3HT-b-PF block copolymers after (a) long-term solvent annealing and (b) thermal annealing above 220 °C. | ||
For thermally annealed samples, melting transitions at roughly 157 °C and 215 °C are detected. These temperatures correspond approximately to the crystal melting temperatures of PF and P3HT, respectively.
DSC measurements thus indicate that the self-assembled lamellar phase melts near 150 °C, while thermally annealed samples exhibit crystal melting temperatures comparable to the constituent homopolymers. Similar results for thermally annealed films have been previously reported for P3HT-b-PF4,21 and other all-conjugated block copolymers.3,18,44–46 For P3HT84-b-PF13, only one thermal transition near 220 °C (near the crystal melting temperature of P3HT) is detected by DSC on both first and second heating cycles, suggesting that PF crystallization is suppressed in this sample due to the much higher content of P3HT in the block copolymer.
Paracrystallinity disorder analysis on lamellar ordering
Peak width analysis can provide information on disorder within crystalline or semicrystalline regions. Disorder is quantified through calculation of a paracrystallinity disorder parameter g,47 calculated using the center (q0) and breadth (Δq) of a peak, as shown in eqn (1).39,48 A larger value of g indicates greater disorder. Here, we present an analysis of g for block copolymer films under different processing histories and for P3HT and PF homopolymer films. The paracrystallinity disorder parameter was measured for the out-of-plane primary lamellar spacing (Fig. 2) in GISAXS and the in-plane π–π stacking reflection (Fig. 5) in GIWAXS in solvent-annealed films. | (1) |
As shown in Table 2, the paracrystallinity disorder parameter g measured for P3HT π–π stacking in thermally-annealed films is in agreement with literature values (g ∼ 6–8%39). The disorder for π–π stacking of the P3HT block is slightly higher for solvent-annealed P3HT-b-PF compared with thermally annealed P3HT homopolymer. This is expected due to connectivity to a PF block which can increase disorder in the P3HT π–π stacking.
| Spacingsa | Samples | g (%) |
|---|---|---|
| a π–π stacking of P3HT and PF was analyzed on GIWAXS patterns whereas 1st lamellar ordering was analyzed on GISAXS patterns. b Commercially available P3HT homopolymer has Mw ∼ 60 kg mol−1. | ||
| π–π stacking, P3HT | P3HT36-b-PF100 | 9.68 |
| P3HT81-b-PF105 | 7.58 | |
| P3HT84-b-PF80 | 9.54 | |
| P3HT84-b-PF13 | 8.42 | |
| Solvent ann. P3HTb | 8.82 | |
| Thermal ann. P3HTb | 8.19 | |
| π–π stacking, PF | P3HT36-b-PF100 | 8.23 |
| P3HT81-b-PF105 | 5.18 | |
| P3HT84-b-PF80 | 8.81 | |
| P3HT84-b-PF13 | 7.59 | |
| Solvent ann. PF | 17.84 | |
| Primary lamellar reflection | P3HT36-b-PF100 | 9.38 |
| P3HT81-b-PF105 | 9.05 | |
| P3HT84-b-PF80 | 9.44 | |
| P3HT84-b-PF13 | 8.89 | |
The paracrystallinity disorder parameter has similar values, roughly 8–10%, for PF π–π stacking and for ordering of the self-assembled lamellae. We also note that PF is more ordered along π–π direction in solvent-annealed BCPs compared with solvent annealed PF homopolymer β phase (g ∼ 17.84%).
Quantitative paracrystallinity disorder analysis thus indicates that ordering along the π–π stacking direction is substantially improved for solvent-annealed films compared with thermally-annealed block copolymers.
Conclusions
A combination of analytical techniques—GIXS, UV-VIS, POM and DSC—reveal novel lamellar ordering in all-conjugated P3HT-b-PF block copolymers after long-term solvent annealing. We observe enhanced π–π stacking in the lamella compared with either the crystal or nematic phases. These results demonstrate that all-conjugated block copolymers exhibit a rich, processing-dependent microstructure, and a quantitative description will have to account for various intermolecular interactions including π–π stacking, steric repulsions of the alkyl side-chains, and chain rigidity. The presence of a liquid crystal phase with enhanced π–π stacking may lead to block copolymer films with superior optoelectronic properties or the use of new processing strategies, such as the application of magnetic fields,13–15 to achieve improved alignment. The results reported for rod–rod all-conjugated block copolymers contrast with previous studies of coil–coil and rod–coil diblock copolymers. In the case of rod–coil block copolymers, self-assembly can be described through a mean field theory that includes Flory–Huggins interactions and Maier–Saupe parameter to account for liquid crystal ordering.17 Here, we observe a qualitatively different self-assembly process driven by both π–π stacking interactions and polymer block segregation.Acknowledgements
This work was supported by the National Science Foundation under Grant no. CBET-1264703 and Louis and Peaches Owen. Research carried out in part at the Center for Functional Nanomaterials and National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract no. DE-AC02-98CH10886. Use of Advanced Photon Source at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract no. DE-AC02-06CH11357. B.S. acknowledges the support of the Department of Homeland Security, Office of Science and Technology, Award # 2009-ST-000031.Notes and references
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS PubMed.
- C. Park, J. Yoon and E. L. Thomas, Polymer, 2003, 44, 6725–6760 CrossRef CAS PubMed.
- M. Sommer, H. Komber, S. Huettner, R. Mulherin, P. Kohn, N. C. Greenham and W. T. S. Huck, Macromolecules, 2012, 45, 4142–4151 CrossRef CAS.
- K. A. Smith, Y.-H. Lin, D. B. Dement, J. Strzalka, S. B. Darling, D. L. Pickel and R. Verduzco, Macromolecules, 2013, 46, 2636–2645 CrossRef CAS.
- C. Guo, Y.-H. Lin, M. D. Witman, K. A. Smith, C. Wang, A. Hexemer, J. Strzalka, E. D. Gomez and R. Verduzco, Nano Lett., 2013, 13, 2957–2963 CrossRef CAS PubMed.
- H. C. Moon, D. Bae and J. K. Kim, Macromolecules, 2012, 45, 5201–5207 CrossRef CAS.
- C. A. Dai, W. C. Yen, Y. H. Lee, C. C. Ho and W. F. Su, J. Am. Chem. Soc., 2007, 129, 11036–11038 CrossRef CAS PubMed.
- B. D. Olsen and R. A. Segalman, Macromolecules, 2005, 38, 10127–10137 CrossRef CAS.
- R. H. Lohwasser, G. Gupta, P. Kohn, M. Sommer, A. S. Lang, T. Thurn-Albrecht and M. Thelakkat, Macromolecules, 2013, 46, 4403–4410 CrossRef CAS.
- S. Y. Choi, J. U. Lee, J. W. Lee, S. Lee, Y. J. Song, W. H. Jo and S. H. Kim, Macromolecules, 2011, 1771–1774 CrossRef CAS.
- Y. J. Lee, S. H. Kim, H. Yang, M. Jang, S. S. Hwang, H. S. Lee and K.-Y. Baek, J. Phys. Chem. C, 2011, 115, 4228–4234 CAS.
- B. McCulloch, G. Portale, W. Bras, J. A. Pople, A. Hexemer and R. A. Segalman, Macromolecules, 2013, 46, 4462–4471 CrossRef CAS.
- M. Gopinadhan, P. W. Majewski, E. S. Beach and C. O. Osuji, ACS Macro Lett., 2011, 1, 184–189 CrossRef.
- M. Gopinadhan, P. W. Majewski, Y. Choo and C. O. Osuji, Phys. Rev. Lett., 2013, 110, 078301 CrossRef.
- H. Tran, M. Gopinadhan, P. W. Majewski, R. Shade, V. Steffes, C. O. Osuji and L. M. Campos, ACS Nano, 2013, 7, 5514–5521 CrossRef CAS PubMed.
- Y.-C. Tung, W.-C. Wu and W.-C. Chen, Macromol. Rapid Commun., 2006, 27, 1838–1844 CrossRef CAS.
- B. D. Olsen, M. Shah, V. Ganesan and R. A. Segalman, Macromolecules, 2008, 41, 6809–6817 CrossRef CAS.
- X. Yu, H. Yang, S. Wu, Y. Geng and Y. Han, Macromolecules, 2012, 45, 266–274 CrossRef CAS.
- R. Verduzco, I. Botiz, D. L. Pickel, S. M. Kilbey, K. Hong, E. Dimasi and S. B. Darling, Macromolecules, 2011, 44, 530–539 CrossRef CAS.
- M. He, F. Qiu and Z. Lin, J. Mater. Chem., 2011, 21, 17039–17048 RSC.
- Y.-H. Lin, K. A. Smith, C. N. Kempf and R. Verduzco, Polym. Chem., 2013, 4, 229–232 RSC.
- U. Scherf, A. Gutacker and N. Koenen, Acc. Chem. Res., 2008, 41, 1086–1097 CrossRef CAS PubMed.
- G. Tu, H. Li, M. Forster, R. Heiderhoff, L. J. Balk, R. Sigel and U. Scherf, Small, 2007, 3, 1001–1006 CrossRef CAS PubMed.
- C. Sinturel, M. Vayer, M. Morris and M. A. Hillmyer, Macromolecules, 2013, 46, 5399–5415 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS PubMed.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu, J. M. J. Fréchet and M. F. Toney, Macromolecules, 2005, 38, 3312–3319 CrossRef CAS.
- S. H. Chen, A. C. Su and S. A. Chen, J. Phys. Chem. B, 2005, 109, 10067–10072 CrossRef CAS PubMed.
- M. Grell, D. D. C. Bradley, G. Ungar, J. Hill and K. S. Whitehead, Macromolecules, 1999, 32, 5810–5817 CrossRef CAS.
- J. Peet, E. Brocker, Y. Xu and G. C. Bazan, Adv. Mater., 2008, 20, 1882–1885 CrossRef CAS.
- B. Lee, I. Park, J. Yoon, S. Park, J. Kim, K.-W. Kim, T. Chang and M. Ree, Macromolecules, 2005, 38, 4311–4323 CrossRef CAS.
- K.-L. Tseng, J. Ruan, Y.-K. Lan, W.-Z. Wang and A.-C. Su, Macromolecules, 2013, 46, 1820–1831 CrossRef CAS.
- E. Verploegen, R. Mondal, C. J. Bettinger, S. Sok, M. F. Toney and Z. Bao, Adv. Funct. Mater., 2010, 20, 3519–3529 CrossRef CAS.
- S. H. Chen, H. L. Chou, A. C. Su and S. A. Chen, Macromolecules, 2004, 37, 6833–6838 CrossRef CAS.
- S. H. Chen, A. C. Su, C. H. Su and S. A. Chen, Macromolecules, 2005, 38, 379–385 CrossRef CAS.
- J. Teetsov and M. Anne Fox, J. Mater. Chem., 1999, 9, 2117–2122 RSC.
- M. Misaki, Y. Ueda, S. Nagamatsu, Y. Yoshida, N. Tanigaki and K. Yase, Macromolecules, 2004, 37, 6926–6931 CrossRef CAS.
- Y.-H. Lin, S. B. Darling, M. P. Nikiforov, J. Strzalka and R. Verduzco, Macromolecules, 2012, 45, 6571–6579 CrossRef CAS.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- B. Grévin, P. Rannou, R. Payerne, A. Pron and J. P. Travers, Adv. Mater., 2003, 15, 881–884 CrossRef.
- M. He, L. Zhao, J. Wang, W. Han, Y. Yang, F. Qiu and Z. Lin, ACS Nano, 2010, 4, 3241–3247 CrossRef CAS PubMed.
- N. Kiriy, E. Jähne, H.-J. Adler, M. Schneider, A. Kiriy, G. Gorodyska, S. Minko, D. Jehnichen, P. Simon, A. A. Fokin and M. Stamm, Nano Lett., 2003, 3, 707–712 CrossRef CAS.
- S. Samitsu, T. Shimomura, S. Heike, T. Hashizume and K. Ito, Macromolecules, 2008, 41, 8000–8010 CrossRef CAS.
- S.-Y. Ku, M. A. Brady, N. D. Treat, J. E. Cochran, M. J. Robb, E. J. Kramer, M. L. Chabinyc and C. J. Hawker, J. Am. Chem. Soc., 2012, 134, 16040–16046 CrossRef CAS PubMed.
- J. Wang, M. Ueda and T. Higashihara, ACS Macro Lett., 2013, 2, 506–510 CrossRef CAS.
- K. Nakabayashi and H. Mori, Macromolecules, 2012, 45, 9618–9625 CrossRef CAS.
- A. M. Hindeleh and R. Hosemann, J. Phys. C: Solid State Phys., 1988, 21, 4155 CrossRef.
- J. Rivnay, R. Noriega, R. J. Kline, A. Salleo and M. F. Toney, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 045203 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, additional GIWAXS and GISAXS images, and POM images of films. See DOI: 10.1039/c3sm53090f |
| This journal is © The Royal Society of Chemistry 2014 |