Ergodic-to-nonergodic phase inversion and reentrant ergodicity transition in DNA–nanoclay dispersions†
Najmul
Arfin
a and
H. B.
Bohidar
*ab
aPolymer and Biophysics Laboratory, School of Physical Sciences, Jawaharlal Nehru University, New Delhi-110067, India. E-mail: bohi0700@mail.jnu.ac.in; Fax: +91 11 2674 1837; Tel: +91 11 2670 4699
bSpecial Centre for Nanosciences, Jawaharlal Nehru University, New Delhi-110067, India
First published on 22nd October 2013
Abstract
We have observed DNA concentration and hydration dependent inversion from ergodic to non-ergodic phase followed by reentry into the ergodic phase in DNA–nanoclay (laponite) dispersions at room temperature (25 °C), using results obtained from dynamic light scattering (DLS) and rheology data. The interaction between the DNA strand and the anisotropically charged discotic platelets of laponite (L) was found to be strongly hierarchical in DNA concentration. For a fixed laponite concentration (CL = 1% (w/v)) and varying DNA concentration (CDNA) from 0.3–2.3% (w/v), we observed three distinct phase regions characterized by the following: region (i): CDNA < 1.0% (w/v), ergodic region with weak DNA–L attractive interaction, region (ii): 1.0% < CDNA < 1.6% (w/v), non-ergodic regime having strong DNA–L associative interaction and region (iii): CDNA > 1.6% (w/v), showing phase reentry into the ergodic regime due to repulsion between DNA strands. Hydration study in these three regions revealed that a loss in the abundance of amorphous water, signified by Raman frequency 3460 cm−1, caused the ergodic to nonergodic phase transition. In summary, it is shown that maximum stability and interaction between DNA and nanoclay platelets occurred at an intermediate concentration of DNA where the hydration was at its minimum. The present system is qualitatively different from the hard-sphere/polymer systems for which reentrant phase transition has been reported in the literature. However, some similarity between the two classes of systems is not ruled out.
I. Introduction
Phase transitions occurring in dispersions of discotic colloidal particles with anisotropic charge distribution has attracted much attention recently. The aging dynamics of these systems yield a rich phase diagram replete with the presence of several phase states that owe their origin to the presence of a hierarchy of time and concentration dependent interactions.1–6 Laponite is a synthetic clay with aspect ratio 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (disc diameter = 30 nm and thickness = 1 nm), which is widely used as a rheology modifier in a variety of industrial products such as paints, varnishes, cosmetics and polymer nanocomposites. For the study of multiple arrested states, charged colloidal clay made of nanometer-sized discotic platelets has emerged as a suitable model material.7–10 The geometric and charge anisotropy associated with these platelets, combined with the presence of attractive and repulsive terms in their interparticle interaction potential, makes the phase diagram of such colloidal systems very rich and complex. In particular, laponite displays nontrivial aging dynamics, replete with multiple arrested states.3–5
:1 (disc diameter = 30 nm and thickness = 1 nm), which is widely used as a rheology modifier in a variety of industrial products such as paints, varnishes, cosmetics and polymer nanocomposites. For the study of multiple arrested states, charged colloidal clay made of nanometer-sized discotic platelets has emerged as a suitable model material.7–10 The geometric and charge anisotropy associated with these platelets, combined with the presence of attractive and repulsive terms in their interparticle interaction potential, makes the phase diagram of such colloidal systems very rich and complex. In particular, laponite displays nontrivial aging dynamics, replete with multiple arrested states.3–5
On the other hand, reentrant phase transition has been observed in a large class of soft matter systems, prominent among them being the colloidal systems. Liu et al.11 have reported reentrant structural phase transition in amphiphilic self-assembly of the quillaja saponin–cholesterol system and here discuss the relevance of precursors for hierarchical structure formation. They have highlighted a three-stage dynamical process for reentrant phase transition caused by cholesterol. Reentrant glass transition in colloid–polymer mixtures with depletion attractions has been reported by Eckert and Bartsch.12 In this experiment, short-range interactions were introduced into a hard-sphere colloidal dispersion by the addition of a polymer that produced novel glass transitions. It was clearly shown that dramatic acceleration of density fluctuations resulted in melting of the colloidal gel. Evidence of reentrant phase behavior in the laponite–polyethylene oxide system was reported by Baghdadi et al.,13 where it was shown that depletion forces caused melting of colloidal glass. In high molecular weight PEO, this glassy phase was reformed. Grandjean and Mourchid14 observed the emergence of a reentrant liquid–glass transition with increasing attraction between soft spheres. They noticed that the particle dynamics exhibited extended logarithmic decay with increasing concentration of sticky soft spheres in the reentrant transition branch. The results were discussed within the framework of mode coupling theory, which stipulates coupling between pairs of density fluctuations. Thus, enhancing the said coupling induced structural arrest and a sharp ergodic-to-nonergodic phase transition. Such phase transitions are an interplay of short and long range forces, entropy effects, structural and charge anisotropy of interacting particles etc.
As far as the reentrant phase transitions of DNA–colloid systems are concerned, Nguyen and Shklovskii15 have reported a detailed study on the interaction profile of DNA and positively charged colloidal macroions and concluded the following: (i) at low colloid concentration, the DNA–colloid complexes are negatively charged, with DNA wrapping the colloids, while (ii) at high colloid concentration the complexes showed charge reversal and revealed a positive charge. The aforesaid two situations were separated by an intermediate phase where the DNA–colloid complexes were fully charge neutralized. Therefore, the concentration of colloidal macroions governed the condensation and reentrant condensation in this system.
A close examination of the literature established that the ergodic-to-nonergodic phase transition has largely been probed by experiments in two classes of systems: colloid–colloid or colloid–polymer. The polymers used were mostly neutral or had low charge density. Herein, we have reported a detail study on the DNA–laponite dispersion and have shown that a high charge density polymer like DNA can hierarchically interact with anisotropically charged laponite platelets to exhibit a ergodic-to-nonergodic phase transition followed by reentry into the ergodic phase. Thus, our system does not belong to any of the aforesaid classes, though some similarities can be invoked, which makes these results unique.
II. Materials and method
Laponite RD was purchased from Southern Clay Products, USA. Laponite powder was used as procured. Laponite RD is a fully synthetic microcrystalline clay with chemical formula Na0.7+[(Si8 Mg5.5Li0.3)O20(OH)4]0.7−. The specific gravity of aponite is 2.53 g cm−3. DNA purchased from Acros Organics having Mw = 50000–100000 Da and almost 300 base pairs was used16 without any further purification. Individual stock solutions were prepared by dispersing laponite and DNA in double distilled deionized water. Samples were prepared by mixing the two dispersions from their stock in the required amount. Samples contained a fixed concentration of laponite (CL = 1% (w/v)), whereas the concentration of DNA (CDNA) was varied between 0 and 2.3% (w/v). No sample was used twice for any measurement. Fresh samples were prepared as per experimental demand. The mixed dispersion had pH value 6.0 ± 0.5. A trace amount of sodium azide was used to avoid degradation of the samples. All experiments were done at room temperature (25 °C) and the relative humidity of the laboratory was close to 50%.
The zeta potential measurements of sample solutions were performed on an electrophoresis instrument (Zeecom – 2000, Microtek, Japan). The zeta potential ξ of a uniformly charged sphere is given by ξ = 4πσ/εκ, where σ is the surface charge density of the particle and ε and κ are the dielectric constant and the Debye–Hückel parameters of the solution, respectively. The surface charge can be determined from the measured zeta potential data if the geometrical shape is known.17 Rheology experiments were performed using an AR-500 model stress controlled rheometer (T. A. Instruments, UK). The elastic moduli of the samples was measured using cone–plate geometry (2 cm diameter, 2° cone angle and 50 mm truncation gap) with the oscillation stress value set at 0.1 Pa. These studies determined the frequency ω dependence of the storage G′(ω) and loss moduli G′′(ω). Rheological characterization of viscoelastic materials is discussed at length by Barnes.18
Dynamic light scattering (DLS) experiments were performed at a scattering angle of 90° and laser wavelength of 632.8 nm on a 256-channel Photocor-FC (Photocor Inc., USA), which was operated in the multi-tau mode (logarithmically spaced channels). The goniometer was placed on a Newport (USA) vibration isolation table. Correlation function for samples at different time intervals were recorded and analyzed for relaxation times. Further details on DLS can be found elsewhere.19 Raman spectroscopy was used to study the hydration behavior of the samples. Raman spectra were recorded on a FT-IR/Raman spectrometer with Microscope – Varian 7000 FTIR, Varian FT-Raman and Varian 600 UMA.
III. Results and discussion
A. Zeta potential of DNA–laponite complex
Electrophoretic measurements assigned the zeta potential values −70 and −27 mV to DNA and laponite, respectively. This attributed a net negative charge to both the components. Electrostatic interaction of DNA with laponite is favored, mainly due to the positively charged edge of laponite (L) binding with DNA. The zeta potential was calculated from the mobility data of the particles streaming in an electric field at low concentration. In an ideal situation, mobility measurements are interpreted assuming the particle to be of spherical geometry, and its concentration is low so as to minimize the effect of inter particle interaction. This enables determination of zeta potential value from the mobility data with relative ease. Fig. 1 clearly shows the binding of DNA to laponite, which causes a decrease in the mobility of complexes formed of the two constituents. This decrease in mobility can be attributed to formation of large and bulky asymmetric complexes. However, it must be realized that we have not yet reached a situation where complete charge neutralization was a distinct possibility, rather it was an artifact of the size of the complex and its geometry. Thus the zeta potential assumed a very low value. Nevertheless, mobility and zeta potential data indicated binding of the positive rim of the laponite platelet to a negatively charged DNA strand. Data were collected under the experimental conditions of a fixed concentration of laponite (CL = 1% (w/v)), and a varying concentration of DNA in the range CDNA = 0.003%–0.6% (w/v) yielded zeta potential values ranging from −27 to −7 mV, thereby implying the partial charge neutralization of the DNA strand due to binding with laponite. A second observation was that binding was strong when CDNA < 0.3% (w/v). Above this concentration the binding persisted, albeit moderately. Such measurements could not be carried out on samples having a higher DNA concentration, due to instrumental limitations. These limitations are based on the fact that measurement of mobility of the samples in the presence of electric field should be done at low concentration so as to avoid the effect of viscous drag. Low concentration measurement gives the mobility of individual particles in an ideal situation and the value of zeta potential with sufficient accuracy. Further investigations were carried out by keeping the concentration of laponite (CL = 1% (w/v)) fixed and varying the DNA concentration (CDNA) from 0.3% to 2.3% (w/v).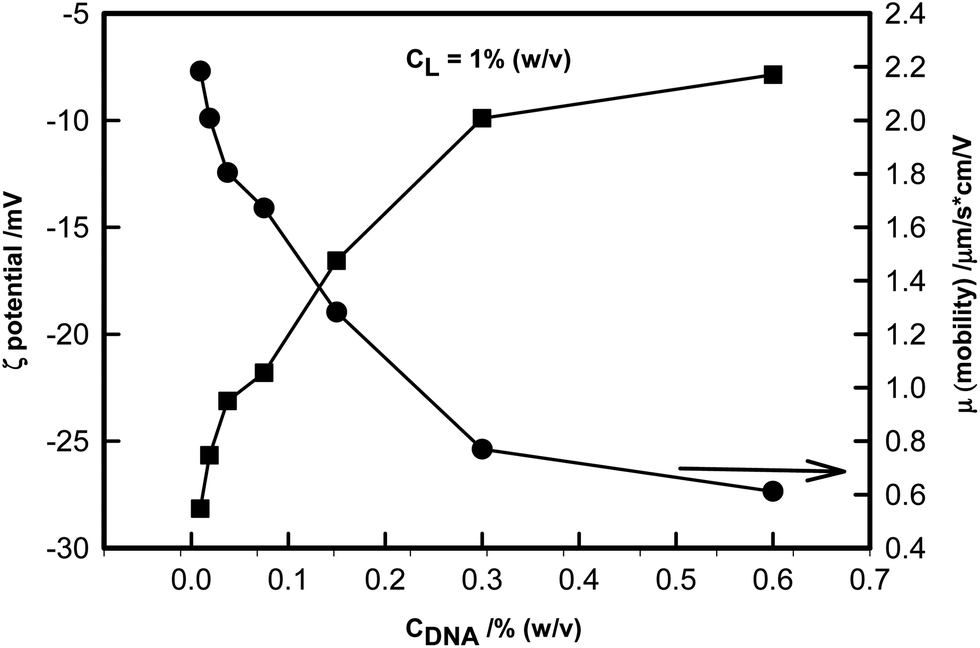 | ||
| Fig. 1 Variation of zeta (ζ) potential and mobility (μ) as a function of DNA concentration shown for fixed CL = 1% (w/v). Note the sharp reduction in zeta potential of the complex due to binding of the edge of the laponite platelet to the DNA strand. | ||
B. Ergodic to nonergodic and reentrant ergodic transition
The samples were subjected to frequency sweep studies to ascertain their viscoelastic properties. The low frequency storage modulus G0, defined as , was determined explicitly, because this parameter provides a quantitative estimate of network rigidity. In our experiments G0 was determined at 0.1 rad s−1. Furthermore, it was noticed that structural evolution of the network was time-dependent. Fig. 2 depicts the dependence of G0 on DNA concentration and with the aging time of the sample. It is clear from Fig. 2 that G0 increased almost by two decades with time (in about 5.5 h) at all DNA concentrations. In region (i), CDNA < 1% (w/v), G0 increased due to the attractive interaction between DNA and laponite. Here, the edge of the laponite disc selectively binds to the DNA strand, causing partial charge neutralization of the nucleic acid (see Fig. 1). This associative interaction saturated in region (ii) between 1.0% < CDNA < 1.6% (w/v), where the maximum attractive interaction prevailed. Further increase in DNA concentration, in region (iii) CDNA > 1.6% (w/v), caused DNA strands in the solution to outnumber the laponite population, leading to strong DNA–DNA repulsion in the solution and hence, G0 started to decrease. Thus the samples in the aforesaid three regions were characterized by viscous, melt-like and viscous attributes, which have been classified as regions (i), (ii) and (iii) respectively.
, was determined explicitly, because this parameter provides a quantitative estimate of network rigidity. In our experiments G0 was determined at 0.1 rad s−1. Furthermore, it was noticed that structural evolution of the network was time-dependent. Fig. 2 depicts the dependence of G0 on DNA concentration and with the aging time of the sample. It is clear from Fig. 2 that G0 increased almost by two decades with time (in about 5.5 h) at all DNA concentrations. In region (i), CDNA < 1% (w/v), G0 increased due to the attractive interaction between DNA and laponite. Here, the edge of the laponite disc selectively binds to the DNA strand, causing partial charge neutralization of the nucleic acid (see Fig. 1). This associative interaction saturated in region (ii) between 1.0% < CDNA < 1.6% (w/v), where the maximum attractive interaction prevailed. Further increase in DNA concentration, in region (iii) CDNA > 1.6% (w/v), caused DNA strands in the solution to outnumber the laponite population, leading to strong DNA–DNA repulsion in the solution and hence, G0 started to decrease. Thus the samples in the aforesaid three regions were characterized by viscous, melt-like and viscous attributes, which have been classified as regions (i), (ii) and (iii) respectively.
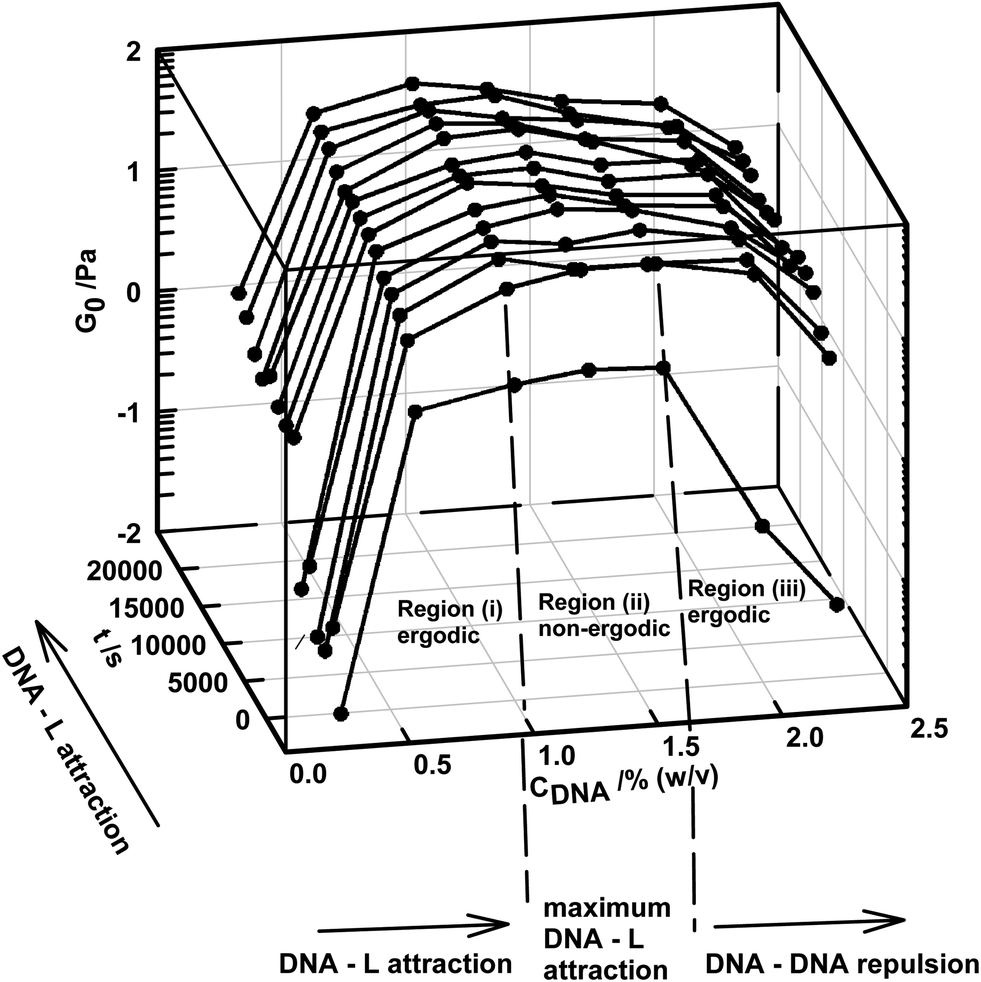 | ||
| Fig. 2 3D plot of elastic moduli at low oscillation frequency (0.1 rad s−1), as a function of CDNA and time for fixed laponite concentration CL = 1% (w/v). Note the evolution of structures with time and DNA content. | ||
The aging behaviour and slow dynamics in soft matter systems is best probed by dynamic light scattering experiments through dynamic structure factor analysis. Usually, the intensity auto-correlation function g2(q, t) is directly correlated to the corresponding dynamic structure factor g1(q, t) through the Siegert relation g2(q, t) = A + B|g1(q, t)|2, where A defines the baseline of the correlation function and |g2(t)|t→∞ = A and B is the spatial coherence factor when the system is ergodic.18 In the arrested phase, scattering centers are localized near their fixed average positions and are only able to execute limited Brownian motion about these positions and non-ergodicity then ensues. A completely ergodic system is one where the scattering moiety is capable of exploring the entire phase space. Furthermore, when the time and the ensemble averages are identical, the system is stationary, implying that the process is independent of the origin of time, which is adequately satisfied only in the case of scattering from homogeneous dilute solutions. The issue of non-ergodicity in DLS experiments has been addressed in several ways, which include rotating the sample cell to scan the entire phase space, expanding the incident beam to collect data from many coherence areas and extracting the non-ergodic contribution from the measured data as a heterodyne contribution.20
Herein, we use the heterodyne approach. The normalized intensity correlation function, g2(q, t), obtained from the sample can be related to the dynamic structure factor, g1(q, t) as
| g2(q, t) = 1 + β′[2X(1 − X)g1(q, t) + X2|g1(q, t)|2] | (1) |
Here, β′ is the coherence factor having a maximum value of 1. In a real experiment, it defines the signal modulation, which is a measure of the signal-to-noise ratio. The parameter X (0 ≤ X ≤ 1) defines the ergodicity through the amount of heterodyne contribution present in the g2(q, t) data. When the value of X = 1, the system is completely ergodic and the Siegert relation is established, whereas in the arrested state X < 1 and the term 2X(1 − X) makes a finite contribution to g2(q, t) and hence it must be accounted for. The intercept of the plot of [g2(q, t) − 1] vs. delay time t at t → 0 gives β′[2X − X2], from which the value of X, which is an instrumental factor, can be calculated if β′ is known. The measured intensity auto-correlation data was analyzed exactly, following the description given elsewhere.21 The pre-factor of the linear term in g1(q, t) in eqn (2) is in most cases much larger than the quadratic second term. Thus
| g1(q, t) ≈ [g2(q, t) − 1]/[2β′(X(1 − X))] | (2) |
The exact evaluation of the dynamic structure factor, g1(q, t), from the measured intensity auto-correlation function g2(q, t) was achieved by substituting the values of β′ and X into eqn (2). A situation may arise where the signal modulation is so low that it is impossible to construct the dynamic structure factor from the measured g2(q, t) data. In such events, the baseline of the evaluated dynamic structure factor provides a quantitative estimation of non-ergodicity, say Y. For a fully ergodic system, the baseline would be located close to Y = 0, whereas for a fully non-ergodic system it will assume a value Y = 1. Thus, in terms of the already defined parameter X, Y = (1 − X).
Typically, the g1(q, t) curve was fitted to a two-exponential decay function defined by two characteristic relaxation times: the fast mode τf and a slow mode τs. This is given by
 | (3) |
Relative amplitudes of the modes are A and B.
Fig. 3a (CDNA = 0.3% (w/v)) depicts region (i) of DNA–L system. Here, single mode relaxation time τ was found to change into two relaxation modes i.e. fast mode relaxation time (τf) and slow mode relaxation time (τs), with sample aging indicating the evolving dynamics in the growth process. However, this region remained in the ergodic phase, as is clear from the indicative correlation curves shown in the inset of Fig. 3a. Fig. 3b (CDNA = 0.6% (w/v)) depicts a deviation in the behavior and the onset of non-ergodicity can be seen at a later stage (inset of Fig. 3b, observe the rising baseline). The relaxation times of both modes begin to slow down and give rise to a single mode relaxation time, with the raised baseline implying approaching dynamic arrest.
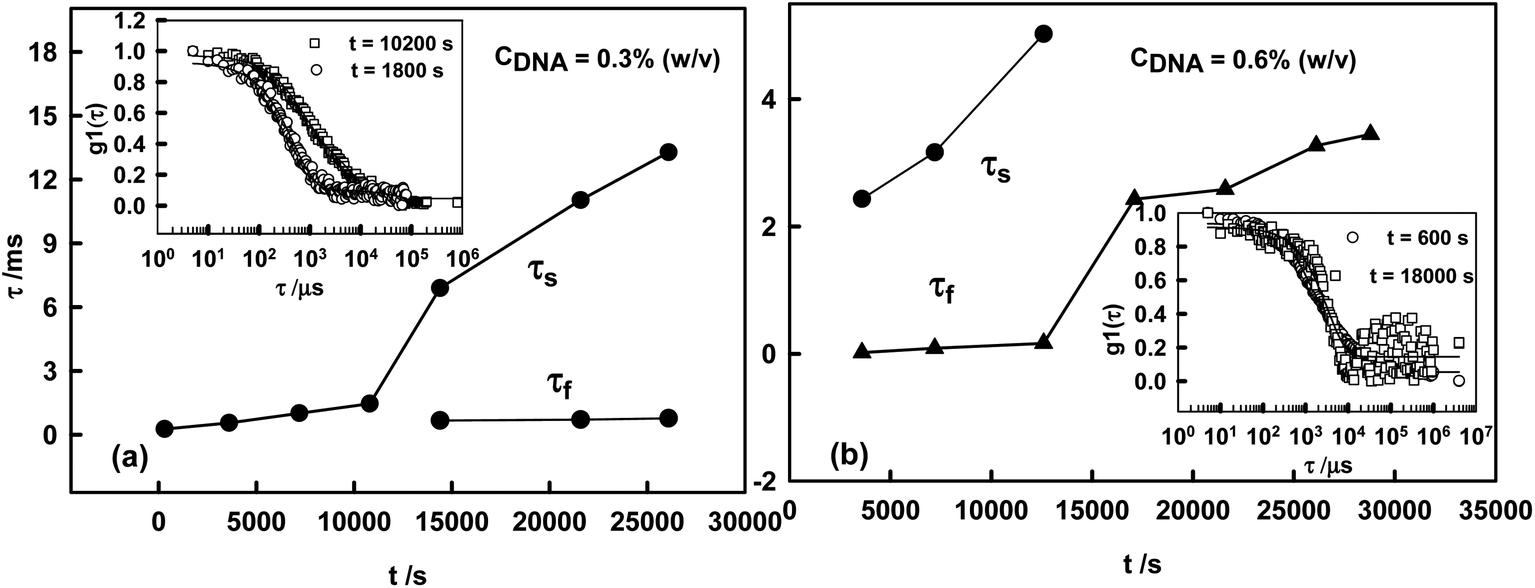 | ||
| Fig. 3 Variation of fast and slow mode relaxation times (τf and τs) as a function of aging time for (a) CDNA = 0.3% (w/v) and (b) CDNA = 0.6% (w/v). The inset shows indicative correlation curves. Note the signature of the onset of non-ergodicity in (b) (inset), indicated by the raised baseline. | ||
Increasing the concentration of DNA led to the system moving deeper into the non-ergodic phase, which is shown in Fig. 4a and b. Data in Fig. 4a (CDNA = 1.0% (w/v)) was fitted to a single exponential function, and it was clearly seen that the relaxation mode (τ) saturated after about 104 s of sample aging, with the system firmly located inside the non-ergodic phase. This non-ergodic behavior can be seen by looking at the indicative correlation functions given in the inset of Fig. 4a, where the baseline is seen to rise considerably.
 | ||
| Fig. 4 Variation of (a) fast relaxation time (τf) at CDNA = 1.0% (w/v) and (b) non-ergodic parameter (Y) at CDNA = 1.3% (w/v), as a function of aging time. Insets show indicative correlation curves. | ||
At CDNA = 1.3% (w/v), the degree of non-ergodicity in the system was so high that we could only register the baselines of correlation curves, as shown in the inset of Fig. 4b, implying a complete dynamic arrest. The baseline analysis of these curves in the time region 105 to 106 μs of decay time yielded the non-ergodicity parameter Y, which increased with aging time, as shown in Fig. 4b. Here, the non-ergodicity parameter is determined from the upward shift of the baseline of correlation curves as discussed earlier.
One interesting observation was made here. The nascent dispersions were associated with non-ergodicity, which increases with aging of the samples. This clearly implies the reorganization of DNA and laponite in the dispersion medium.
Further increase of CDNA to 1.6% (w/v) raised abundance of DNA strands, where enhanced DNA–DNA repulsion caused reorganization of the intermolecular complexes. The mobility of the complexes started to increase, which led to the transition from the non-ergodic to the ergodic phase. Analyzing the baseline of the correlation curves (inset Fig. 5a), in the region 105 to 106 μs of decay time, we observed that non-ergodicity parameter Y decreased with time, as shown clearly in Fig. 5a. At CDNA = 2.3% (w/v), ergodicity was restored completely, which is clear from the indicative correlation curves shown in the inset of Fig. 5b. Fig. 5b depicts a slight decrease in the fast and slow relaxation mode relaxation times with aging, which is a signature of the transition from the non-ergodic to the ergodic phase. Thus, we have clearly observed reentrant ergodic transition in our system fully driven by DNA concentration.
 | ||
| Fig. 5 Variation of (a) ergodic parameter (Y) at CDNA = 1.6% (w/v) and (b) fast and slow mode relaxation times at CDNA = 2.3% (w/v), as a function of sample aging time. The inset shows indicative correlation curves. Notice the system getting out of the trapped state due to reentry into the ergodic phase with aging. | ||
Analysis of DLS data supported the rheology results to a large extent. DLS classified region (i) as the ergodic region, where a weak DNA–L attraction existed (Fig. 3a and b) and DNA–L complexes could explore the entire phase space. Region (ii) is classified as a non-ergodic region where the correlation function ceased to exist due to immobility of the DNA–L complex (Fig. 4a and b). In region (iii), reentrant ergodicity behavior owing to preferential DNA–DNA repulsion was noted (Fig. 5a and b). A simplistic model, which describes the interaction between DNA and laponite, has been formulated and discussed in some detail (see ESI†).
C. Deformation of elastic network
Fig. 6 depicts the viscoelastic parameters of the system at various values of CDNA, obtained from isothermal frequency sweep experiments carried out at room temperature. Frequency sweep studies have been done in oscillatory mode at constant oscillation stress, which provided information about the microstructure. Rheology provides explicit information about the storage/elastic modulus G′(ω), thereby probing the elastic and network property of the materials completely.22 It is clear from Fig. 6 that the value of G′(ω) increased with increasing DNA concentration in the range CDNA = 0.3–1.3% (w/v), and then decreased in the range CDNA = 1.6–2.3% (w/v) for the same time frame. However, G′(ω) increased irrespective of DNA concentration over a period of time at all CDNA, which clearly implied evolving network formation between DNA and laponite with time. The explicit frequency dependence of the complex modulus was determined by fitting the data to the following power-law function| G′(ω) ∼ ωn | (4) |
 | ||
| Fig. 6 Frequency sweep of elastic moduli for CDNA (a) 0.3%, (b) 0.6%, (c) 1.0%, (d) 1.3%, (e) 1.6%, (f) 2.0% and (g) 2.3% (w/v) at different time intervals. The data were fitted to eqn (4). | ||
The linear visco-elasticity model, for pre and post gel situations, proposed by Winter and Chambon,23 predicts that the stress-relaxation will follow the power-law frequency dependence behaviour given by eqn (4) with 0 < n < 1. Stoichiometrically balanced and imbalanced crosslinked networks showed n = 1/2 (excess crosslinker) and n > 1/2 (lack of crosslinker), respectively. However, this description strictly applies to chemically crosslinked gels. Here, the very low values obtained for n indicate the elasticity of the materials. The elastic strength increased with increasing CDNA and the maximum strength found was ≈80 Pa at 122 h in the system with CDNA = 1.3% (w/v). The elastic strength was reduced on addition of more DNA in the dispersion, due to intermolecular repulsion between the DNA strands.
Fitting to frequency sweep data for the time period 7–122 h was carried out as per eqn (4), as shown in Fig. 6. The values of n are plotted as a function of CDNA in Fig. 7, where these values were calculated for the samples concerned after allowing aging for 122 h. Fig. 7 illustrates clearly that after CDNA = 1.6% (w/v), the value of n increased. This increase denotes reorganization of the network structure, which owes its origin to DNA–DNA repulsion.
 | ||
| Fig. 7 Variation of the exponent n determined from eqn (4) as a function of DNA concentration. Note the sharp rise in the value of the exponent in region (iii), signifying strong DNA–DNA repulsion. The line is a guide to the eye, and the arrows demarcate various viscoelastic regions. See text for details. | ||
D. Effect of hydration
Raman spectroscopy has been used extensively in the literature to address the issue of hydration in complex soft matter systems. The information obtained from such data indicates the hydration of DNA–L complexes. Thus, it provides a suitable foundation for discussion of the solvation behaviour of the moiety concerned. Herein, Raman spectra were used to investigate the hydration behavior of the DNA–L system, as this technique is very sensitive to the vibrational mode of the O–H bond. The peaks located at 3200, 3310 and 3460 cm−1 are related to O–H vibrational modes of structured water, partially structured water and amorphous water, respectively. The relative contributions of these three vibrational modes are shown in Fig. 8. Two key observations could be made from this data: (i) at the concentration of maximum interaction (CDNA = 1.0% (w/v)) there was a small gain (10%) in the availability of structured water (3200 cm−1 band), and (ii) both in regions (i) and (iii) the presence of amorphous water (3460 cm−1) was close to 40% (system in ergodic state). In contrast, in region (ii), the non-ergodic state, there was a severe depletion in the availability of this type of water, with the amount dropping to less than 25%. Correspondingly, the propensity of partially structured water increased from 40 to 60%. Hence, the hydration was largely provided by the amorphous water, a conjecture largely supported by the Raman data shown in Fig. 8. Thus, it can be concluded that loss of hydration drove the system from the ergodic to the nonergodic state. Such an observation was not reported hitherto. | ||
| Fig. 8 Variation of the fractional area of Raman bands corresponding to O–H vibrations: (A) 3200 and 3310 cm−1 and (B) 3460 cm−1 as a function of CDNA. The fractional area is indicative of the abundance of a specific type of water. The lines are guide to the eye and the arrows demarcate various hydration regions. See text for details. | ||
It has been observed that the biological function of proteins and other intracellular biopolymers cannot be understood without taking into account their hydration behaviour.24 In addition, the ionic state of the aqueous solution has considerable influence on the biological function of biomolecules.25 Furthermore, the mobility of water on inter and intra molecular length scales, and the effect of differential hydration features, have been reported for different soft matter systems.26–28 In the current study, we have clearly observed the correlation between hydration and ergodicity of a given soft matter phase.
IV. Summary
Interaction between DNA and laponite has been studied, which revealed that the interaction between the two was hierarchical in DNA concentration. The interaction between the two components was largely electrostatic in nature. Three distinct regions of interaction, categorized as ergodic, non-ergodic and reentrant ergodic, were clearly observed. It was also observed that the samples exhibited characteristic viscoelastic signatures in their ergodic and nonergodic phase states. All the samples revealed a strong aging effect, with their network strength increasing with time. It was observed that the amorphous water, which provided hydration to the complexes, depleted severely in the nonergodic region. When this hydration was restored to its initial value (40%), reentrant ergodic transition was revived. Thus, the ergodic to nonergodic transition was caused by the loss in hydration, an observation not reported hitherto. In summary, it is concluded that hydration data determined from Raman spectroscopy and viscoelastic information deduced from rheology could clearly define the ergodicity of various phase states and their evolution with aging.Acknowledgements
N. Arfin acknowledges receiving a Senior Research Fellowship from Council for Scientific and Industrial Research, Government of India and AIRF (JNU) for use of the Raman facility.References
- B. Ruzicka, E. Zaccarelli, L. Zulian, R. Angelini, M. Sztucki, A. Moussaïd, T. Narayanan and F. Sciortino, Observation of empty liquids and equilibrium gels in a colloidal clay, Nat. Mater., 2011, 10, 56–60 CrossRef CAS PubMed.
- E. Zaccarelli, Colloidal gels: equilibrium and non-equilibrium routes, J. Phys.: Condens. Matter, 2007, 19, 323101–323150 CrossRef.
- S. C. Glotzer and M. J. Solomon, Anisotropy of building blocks and their assembly into complex structures, Nat. Mater., 2007, 6(8), 557–562 CrossRef PubMed.
- A. Knaebel, M. Bellour, J. P. Munch, V. Viasnoff, F. Lequeux and J. L. Harden, Aging behavior of laponite clay particle suspensions, Europhys. Lett., 2000, 52, 73–79 CrossRef CAS.
- B. Abou, D. Bonn and J. Meunier, Aging dynamics in a colloidal glass, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2001, 64, 021510–021516 CrossRef CAS.
- S. Jabbari-Farouji, G. H. Wegdam and D. Bonn, Gels and Glasses in a Single System: Evidence for an Intricate Free-Energy Landscape of Glassy Materials, Phys. Rev. Lett., 2007, 99, 065701–065704 CrossRef.
- S. Jabbari-Farouji, G. H. Wegdam and D. Bonn, Ageing dynamics of translational and rotational diffusion in a colloidal glass, J. Phys.: Condens. Matter, 2004, 16, 471–477 CrossRef.
- B. Ruzicka, L. Zulian and G. Ruocco, More on the phase diagram of laponite, Langmuir, 2006, 22, 1106–1111 CrossRef CAS PubMed.
- B. Ruzicka and E. Zaccarelli, A fresh look at the laponite phase diagram, Soft Matter, 2011, 7, 1268–1286 RSC.
- B. Ruzicka, L. Zulian, E. Zaccarelli, R. Angelini, M. Sztucki, A. Moussaïd and G. Ruocco, Competing Interactions in Arrested States of Colloidal Clays, Phys. Rev. Lett., 2010, 104, 085701–085704 CrossRef CAS.
- J. Liu, M. Harms, V. M. Garamus and C. C. Muller-Goymann, Reentrant structural phase transition in amphiphilic self-assembly, Soft Matter, 2013, 9, 6371 RSC.
- T. Eckert and E. Bartsch, Re-entrant glass transition in a colloid–polymer mixture with depletion attractions, Phys. Rev. Lett., 2002, 89, 125701–125704 CrossRef CAS.
- H. A. Baghdadi, E. C. Jensen, N. Easwar and S. R. Bhatia, Evidence for re-entrant behavior in laponite–PEO systems, Rheol. Acta, 2008, 47, 121–127 CrossRef CAS PubMed.
- J. Grandjean and A. Mourchid, Re-entrant glass transition and logarithmic decay in a jammed micellar system. Rheology and dynamics investigation, Europhys. Lett., 2004, 65(5), 712–718 CrossRef CAS.
- T. T. Nguyen and B. I. Shklovskii, J. Chem. Phys., 2001, 115, 7298–7308 CrossRef CAS.
- N. Arfin and H. B. Bohidar, Condensation, complex coacervation and overcharging during DNA–gGelatin interactions in aqueous solutions, J. Phys. Chem. B, 2012, 116, 13192–13199 CrossRef CAS PubMed.
- H. Ohshima, Adv. Colloid Interface Sci., 1995, 62, 189 CrossRef CAS.
- H. A. Barnes, Handbook of Elementary Rheology, University of Wales Press, Wales, 2000 Search PubMed.
- B. J. Berne and R. Pecora, Dynamic Light Scattering with Applications to Chemistry, Biology and Physics, Wiley-Interscience, New York, USA, 1976 Search PubMed.
- T. Coviello, E. Geissler and D. Meier, Macromolecules, 1997, 30, 2008 CrossRef CAS.
- R. K. Pujala and H. B. Bohidar, Ergodicity breaking and aging dynamics in laponite–montmorillonite mixed clay dispersions, Soft Matter, 2012, 8, 6120–6127 RSC.
- J. D. Ferry, Viscoelastic Properties of Polymers, John Wiley, New York, 1961 Search PubMed.
- H. H. Winter and F. J. Chambon, Analysis of linear viscoelasticity of a crosslinking polymer at the gel point, J. Rheol., 1986, 30, 367–382 CrossRef CAS.
- P. Ball, Water as an Active Constituent in Cell Biology, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- J. T. Trevors and G. H. Pollack, The origin of life in a hydrogel environment, Prog. Biophys. Mol. Biol., 2005, 89, 1–8 CrossRef CAS PubMed.
- A. Y. Grosberg, T. T. Nguyen and B. I. Shoklovskii, Colloquium: The physics of charge inversion in chemical and biological systems, Rev. Mod. Phys., 2002, 74, 329–345 CrossRef CAS.
- T. Seydel, L. Wiegart, F. Juranyi, B. Struth and H. Schober, Unaffected microscopic dynamics of macroscopically arrested water in dilute clay gels, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 061403–061408 CrossRef.
- J. Israelachvili and H. Wennerstrom, Role of hydration and water structure in biological and colloidal interactions, Nature, 1996, 379, 219–225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A simplistic model depicting the interaction between DNA and laponite has been formulated and discussed. See DOI: 10.1039/c3sm52218k |
| This journal is © The Royal Society of Chemistry 2014 |