 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Arylsulfanyl radical lifetime in nanostructured silica: dramatic effect of the organic monolayer structure†
François
Vibert
a,
Sylvain R. A.
Marque
a,
Emily
Bloch
b,
Séverine
Queyroy
a,
Michèle P.
Bertrand
*a,
Stéphane
Gastaldi
*a and
Eric
Besson
*a
aAix-Marseille Université, CNRS, Institut de Chimie Radicalaire UMR 7273, 13397 Cedex 20, Marseille, France. E-mail: stephane.gastaldi@univ-amu.fr; eric.besson@univ-amu.fr
bAix-Marseille Université, CNRS, MADIREL UMR 7246, 13397 Cedex 20, Marseille, France
First published on 4th July 2014
Abstract
Nanostructured hybrid silicas, in which covalently anchored aromatic thiols are regularly distributed over the pores, enable a dramatic increase in the half-lives of the corresponding arylsulfanyl radicals. This enhancement is not only due to limited diffusion but also to the structure of the organic monolayer on the surface of the pores. Molecular dynamics modeling shows that at high loadings, in spite of their spatial vicinity, supramolecular interactions disfavor the coupling of arylsulfanyl radicals. As compared to phenylsulfanyl radical in solution, the half-life measured at 293 K can be increased by 9 orders of magnitude to reach 65 h.
Development of new materials with unusual functional groups, such as transient radicals, is of topical interest to enable new advances in spin sciences. The notions of transience, persistence and stability of radical species are not widespread amongst the chemical community. Radicals can be classified depending on their lifetime. According to Ingold, a radical can be transient (half-life < 10−3 s), persistent (half-life > 10−3 s) or even stable (storable in air).1,2 Therefore, increasing the half-life of a radical species from a microsecond to a tenth of second can be considered as making persistent a transient radical. Most radicals are highly reactive and transient in nature. The propensity of unpaired electrons to pair up with one another to form closed-shell molecules is responsible for the high rate constants of radical/radical reactions and of most radical/molecule reactions.3 However, many radicals, such as for instance triphenylmethyl, phenalenyl, hydrazyl, nitroxyl or thioaminyl radicals, transgress this behavior.4 These radicals can be tailored to become extremely persistent or even stable through the subtle interplay of steric and/or electronic effects.
Stable open shell molecules are useful tools. They are extensively applied in electron paramagnetic resonance (EPR) spectroscopy techniques including spin trapping,5–8 spin labeling9 or EPR imaging.10 Other properties related to the presence of unpaired electrons, such as conductivity or magnetism, have been studied in order to develop smart devices.11,12 Further progress in spin sciences, based on stable neutral radicals, can be expected from the design and synthesis of stable neutral radicals with novel molecular and electronic structures.13 In this context, the development of a versatile approach enabling the transformation of a transient radical to a highly persistent or stable one without changing its structural features remains a challenging task. This can lead to the discovery of new properties of radical species, which cannot be explored at present due to their rapid decay.
Organic radicals and materials already have a long common history, as evidenced by the pioneering studies performed at low temperature in the 1960s.14 More recently, organic radical precursors have been adsorbed in zeolites.15–19 Extensive studies of the influence of supramolecular binding with zeolites on the recombination of benzylic radicals have been reported by Turro, who correlated the rate of radical decay to steric effects in internal supercages. Organic radical precursors have also been grafted onto silica20 and nanostructured silicas.21–23 The increase of radical lifetimes,15,20 as well as the modification of their reactivity,21 was rationalized in terms of limited diffusion and/or interactions with the surface. It must be underlined that, in all these studies, the organic radical precursor was introduced into the material by simple adsorption or by post-grafting, in other words with no means to control the distribution of the radical precursor in the inorganic material.
We report herein a versatile approach to increase the persistence of radicals by the simultaneous control of the diffusion and the close environment of the radical species, and not by the modification of steric or electronic factors. To reach this goal, radical precursors must be accurately located in an inorganic matrix, designed to prevent termination reactions. The direct synthesis of organic–inorganic hybrid nanostructured materials, by the use of the sol–gel process in the presence of structure-directing agents, enables functional nanostructured silicas in which the radical precursors are regularly distributed in the pores to be engineered.24 This approach offers the possibility to investigate the influence of the structure of the hybrid material on the radical lifetime and, as a consequence, to correlate the persistence to parameters such as loading, nature of the spacer, and presence of residual silanols on the pore surface.
Results and discussion
To implement such an approach, a model radical, showing a broad range of reactivity in solution, was selected. Arylsulfanyl radicals were considered good models since they are reactive species involved in hydrogen atom abstraction, addition to π systems or dimerization reactions.25–27 They are, by nature, transient, as illustrated by their diffusion-controlled recombination rate.28 They can be easily generated by photolysis of the corresponding thiophenols.Although arylsulfanyl radicals are transient species, they have been extensively studied and characterized by a large number of spectroscopic methods such as EPR or laser flash photolysis.26,29 The EPR signal of phenylsulfanyl radical has been detected in organic glasses.26,30 From flash photolysis, its half-life was estimated as about 200 μs at room temperature.31 We found that a signal could only be observed below 153 K upon irradiation of a thiophenol benzene glass. Moreover, when the same experiment was reproduced with thiophenol adsorbed on non-functionalized SBA-15 silica, the spectrum of the sulfanyl radical was observed below 173 K and no persistence was noted. There is no effect of the nanostructured environment on the lifetime of phenylsulfanyl radical.
Besides the ease of generating radicals from UV-transparent hybrid silicas by simple photolysis, the use of an arylthiol as a radical precursor leads to several advantages that will be discussed below.
Triethoxysilane derivatives 1 and 2 were selected as radical precursors (Scheme 1). The thiol group was protected as a thioester in order to avoid the formation of disulfides during the synthesis of the hybrid material. The hydrolysis of the thioester groups during the sol–gel process generated thiol functions, which led to anchored arylsulfanyl radicals upon UV irradiation. The main difference between 1 and 2 lies in the chemical nature of the spacer connecting the aromatic ring to the surface of the silica, i.e., urea and ether, respectively. Assuming a fully extended molecule, the distances between silicon and sulfur atoms in the most stable conformation were estimated (B3LYP/6-31+G(d,p)) to be 1.36 and 1.11 nm for the thiophenols corresponding to 1 and 2, respectively.
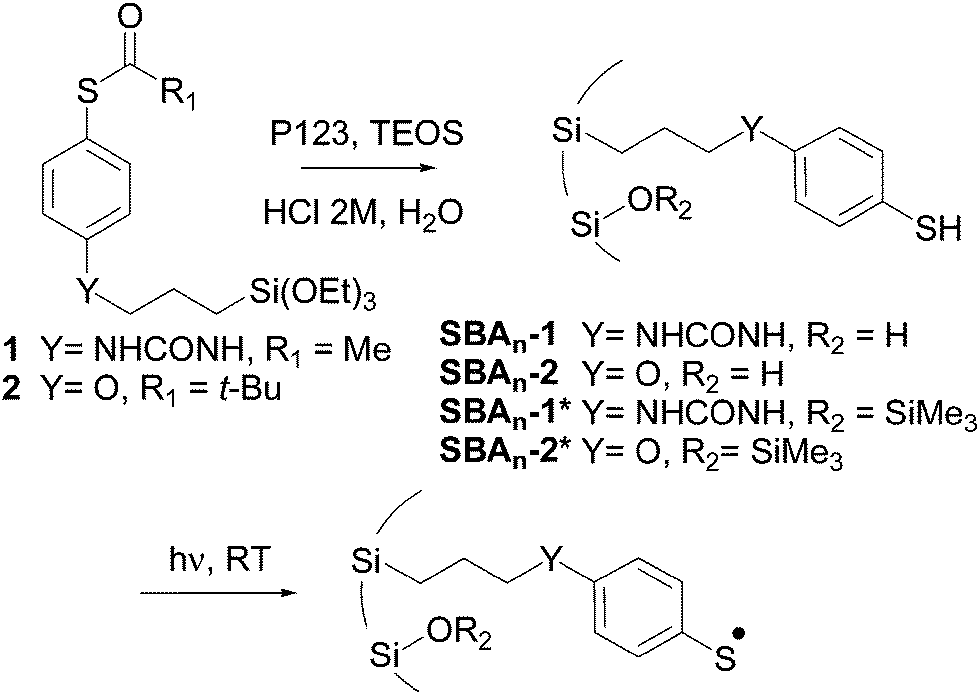 | ||
| Scheme 1 Synthesis and photolysis of radical precursors localized in the pores of the silica. | ||
The first step in our approach consisted of the preparation of a series of periodic mesoporous hybrid silicas in which the radical precursors were covalently linked to silicon atoms and localized inside the pores. 2D hexagonal SBA type silicas were prepared by co-condensation of alkyltriethoxysilane 1 or 2 and tetraethyl orthosilicate (TEOS) in the presence of P123 (PEO20PPO70PEO20) as a structure-directing agent. The resulting materials were named SBAn-1, in which 1 specifies the nature of the organic precursor and n indicates the TEOS/radical precursor molar ratio determined after the characterization of the hybrid silicas. The use of the sol–gel process in the presence of structure-directing agents induces a regular distribution of the organic radical precursor. The average distance between organic chains was estimated from standard analyses (Table 1).
| Entry | g ‖ | g ⊥ | g iso | d | t 1/2 (h) | |
|---|---|---|---|---|---|---|
| a g iso = (2g⊥ + g‖)/3. b Statistical average distance between organic precursors in nm. c Determined from the double integrated EPR signal at 293 K. d Not determined. | ||||||
| a | SBA17-1 | 2.0141 | 2.0066 | 2.0091 | 1.2 | >62.4 |
| b | SBA33-1 | 2.0143 | 2.0050 | 2.0081 | 1.6 | >15.0 |
| c | SBA71-1 | 2.0155 | 2.0051 | 2.0086 | 4.6 | >3.6 |
| d | SBA239-1 | 2.0146 | 2.0060 | 2.0089 | 19.6 | >18.0 |
| e | SBA21-2 | 2.0142 | 2.0077 | 2.0099 | 0.9 | ndd |
| f | SBA37-2 | 2.0142 | 2.0057 | 2.0085 | 1.9 | 9.5 |
| g | SBA73-2 | 2.0150 | 2.0054 | 2.0086 | 4.8 | 3.2 |
As mentioned above, deacetylation of precursors 1 and 2 occurred during the synthesis of the hybrid silicas. This was confirmed by the disappearance of the thioester carbonyl signal in 13C CP-MAS NMR spectra at 194.6 and 200.3 ppm for 1 and 2, respectively.
The behavior of arylsulfanyl radicals generated from precursors regularly anchored inside the pores of the silicas was studied by EPR. In a typical procedure, the functionalized material was degassed (10−5 mbar) in a 4 mm quartz-glass tube and arylsulfanyl radicals were generated via the photolytic cleavage of the S–H bonds inside the spectrometer cavity (xenon lamp (200–800 nm)) (Scheme 1).
An anisotropic signal, resulting from the immobilization of the precursors on the surface of the pores, was recorded regardless of the hybrid silica and the loading. The EPR parameters of these signals were in agreement with the formation of arylsulfanyl radicals,32–34 for which the g tensor is axisymmetrical and the spin density is localized on the sulfur atom.35,36 Representative spectra are shown in Fig. 1.
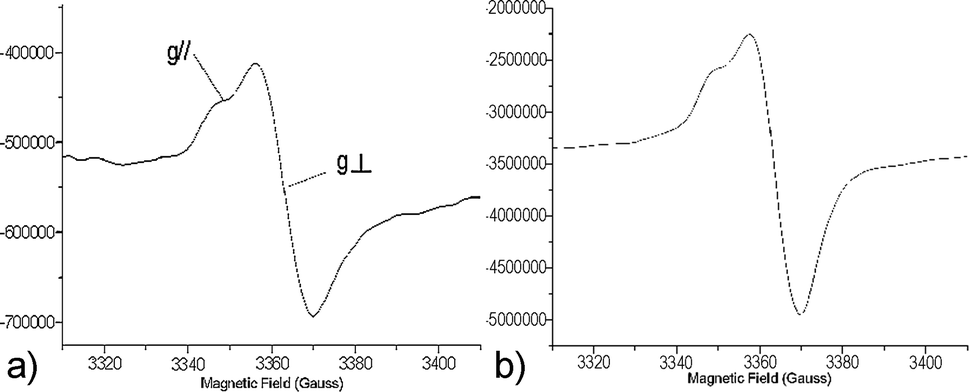 | ||
| Fig. 1 EPR spectra at 293 K for the irradiation of SBA33-1 (a) and SBA37-2 (b). | ||
The radical lifetimes are collected in Table 1. For both SBAn-1 and SBAn-2, the measured giso37 ranged from 2.0081 to 2.0099.
It must be underlined that, regardless of the structure of the organic linker, SBAn-1 or SBAn-2, a low intensity signal corresponding to arylsulfanyl radicals was observed before irradiation. This might result from the homolysis of a small amount of precursor under daylight. In all experiments, the irradiation was maintained until the signal reached a quasi-stationary maximum intensity, at which point the UV lamp was switched off. The half-lives of the anchored arylsulfanyl radicals were measured after turning off the irradiation by monitoring the decay of the signal intensity (determined from the double integrated EPR signal). The decay curves were fitted with exponential or multi-exponential models, which did not allow for the determination of the mechanism involved. Several termination processes occurred, and the half-lives were graphically estimated from the decay curves.
A dramatic increase in the lifetime compared to the value reported for phenylsulfanyl radical in solution, i.e. 200 μs,31 was recorded in each case.
– Concerning the half-life (t1/2) of the radicals resulting from the irradiation of SBAn-1, the highest value, i.e. greater than 62.4 h, was recorded for the highest concentration of organic precursor, i.e.SBA17-1. One then observed a decrease in lifetime with loading. Half-lives greater than 15 h and 3.6 h were registered for SBA33-1 and SBA71-1, respectively. In the case of the highest dilution, SBA239-1, t1/2 exceeded 18.0 h.
– Regarding the material with the oxygenated linker, an intense EPR signal was recorded for SBA21-2 prior to irradiation. The increase of the signal was weak under irradiation. Similarly, its decrease was weak after the end of the photolysis. This phenomenon might be due to the interference of spontaneous or photo-stimulated electron transfer reactions.38 Even though this signal indicated the presence of a persistent radical species, it was difficult to determine its half-life. For lower loadings, SBA37-2 and SBA73-2, the measured t1/2 values were 9.5 and 3.2 h, respectively.
In order to determine the effect of the surface residual silanol groups on the behavior of the radicals, the hybrid silicas were passivated upon reaction with ClSiMe3 (Scheme 1). Photolytic EPR experiments were repeated with these new hybrid materials, SBAn-1* and SBAn-2* (Table 2).
| Entry | g ‖ | g ⊥ | g iso | t 1/2 (h) | |
|---|---|---|---|---|---|
| a g iso = (2g⊥ + g‖)/3. b Determined from the double integrated EPR signal at 293 K. c Loss of 32% of signal intensity in 80 h. | |||||
| a | SBA17-1* | 2.0141 | 2.0053 | 2.0082 | >65.0 |
| b | SBA33-1* | 2.0149 | 2.0038 | 2.0075 | >63.6 |
| c | SBA71-1* | 2.0145 | 2.0043 | 2.0077 | 8.2 |
| d | SBA239-1* | 2.0139 | 2.0056 | 2.0084 | 16.4 |
| e | SBA21-2* | 2.0136 | 2.0078 | 2.0097 | ndc |
| f | SBA37-2* | 2.0141 | 2.0079 | 2.0100 | 62.9 |
| g | SBA73-2* | 2.0141 | 2.0061 | 2.0088 | 3.8 |
Once again, except for SBA17-1*, a huge improvement in t1/2 was observed compared to phenylsulfanyl radical in solution, and the passivation accentuated this enhancement. This passivation increased the half-life by 48.6 h for SBA33-1* as compared to the corresponding non-passivated materials. A half-life greater than 63.6 h was recorded for SBA33-1*. The passivation reduced the difference observed between SBA17-1 and SBA33-1. The half-life was more than 2 times higher for SBA71-1* compared to SBA71-1. The same passivation effect was also noted with SBA37-2* and SBA73-2*, where the half-lives reached 62.9 and 3.8 h, respectively. Because of its dramatic increase, t1/2 was not measured in the case of SBA21-2*; the signal lost only 32% of its intensity over 80 h. In other words, the half-life was by far the highest of all the series. In the case of SBA17-1*, the limited effect of the passivation might be explained by the low number of silanols available for the passivation due to the loading.
In such a system, the two main processes by which arylsulfanyl radicals can evolve are dimerization and inter-chain hydrogen atom transfer (HAT). A significant interest in selecting thiols as radical precursors is that the degenerate HAT from an S–H group to a sulfur-centered radical does not contribute to the disappearance of arylsulfanyl radicals.
As the spin density is mainly localized on the sulfur atom, recombination forming disulfides is the most likely cause of the decay of the arylsulfanyl radical signals.35 MP2 calculations show that, in the cases of the 4-methoxy or the 4-methylureido phenylsulfanyl radical, 85% of the spin density lies on the sulfur atom. Remarkably, the data collected in Tables 1 and 2 showed that the closer the organic precursors, the higher the radical lifetimes. This behavior is counter-intuitive, since one would have expected that the closer the radicals are, the easier their dimerization should be, resulting in shorter lifetimes.
There is formally no activation energy for the coupling of two freely diffusing phenylsulfanyl radicals. Calculations did not allow the location of a transition structure.39 Disulfides are known to adopt a preferential skew conformation.40 Experimentally, a 85° dihedral angle has been measured for crystalline diphenyl disulfides.41–43 This value is coherent with our own calculations at the B3LYP/6-311+G(d,p) level, which predict an 82.5° dihedral angle (CSSC), a 104° (CSS) valence angle and a 2.13 Å S–S bond length (Fig. 2), and with MP2 calculations,36 which predict a dihedral angle of 80°. This conformation enables the repulsion between the sulfur lone pairs to be minimal. The conformer is stabilized by the hyperconjugative interactions between the lone pair on one sulfur atom and the antibonding σ*S–C orbital on the other sulfur atom, which increase the bond order of the S–S bond. The stability and the reactivity of the S–S bridge depend on the mechanical forces exerted by its environment.44,45 Disulfur bridges can undergo spontaneous and reversible cleavage. The biradical character of the S–S bond increases upon stretching.46,47 It is well known that torsional strain also plays a key role: compressing the dihedral angle down to 0° enhances the 4 electron destabilization. The resulting increase of the potential energy is amplified by 1,4-steric repulsions in the cis conformation.
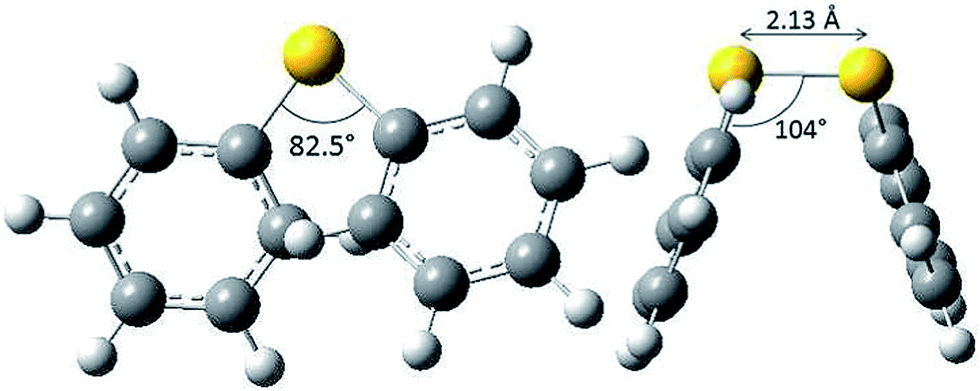 | ||
| Fig. 2 Favored conformation of diphenyldisulfide (B3LYP/6-311+G(d,p) level of theory). | ||
On the basis of these data, and assuming that the formation of the disulfide linkage could only be envisaged between two spatially close arylsulfanyl radicals, we have investigated the behavior of organic moieties in the pore of the silica by applying classical molecular dynamics to the modeling of an isolated functionalized pore. This simulation method is neither able to study the coupling reaction nor its reversal. However, its capacity to model large size systems leads to interesting conformational analyses. The model cylinders of silica (inner diameter 5 nm, length 6.4 nm) were built according to the Brodka approach48 and randomly functionalized with the arylsulfanyl radicals (CH2–CH2–CH2–O–C6H4–S˙) derived from 2. Three virtual hybrid silicas were prepared with average inter-chain distances of 0.56, 1.10 and 1.75 nm, and were named vSBAA-2, vSBAB-2 and vSBAC-2, respectively. These systems were simulated under two different force fields in order to exclude any bias in the results due to the choice of one set of parameters.
Modeling put into perspective the size of the pore in comparison with that of the organic precursors (Fig. 3a, c and e). The following interpretation of the data is based on the reasonable assumption that, in such a system, an arylsulfanyl radical can mainly disappear by coupling with another radical in close vicinity. In addition, the disulfide linkage should be formed in a stable conformation with minimal external mechanical stress.44
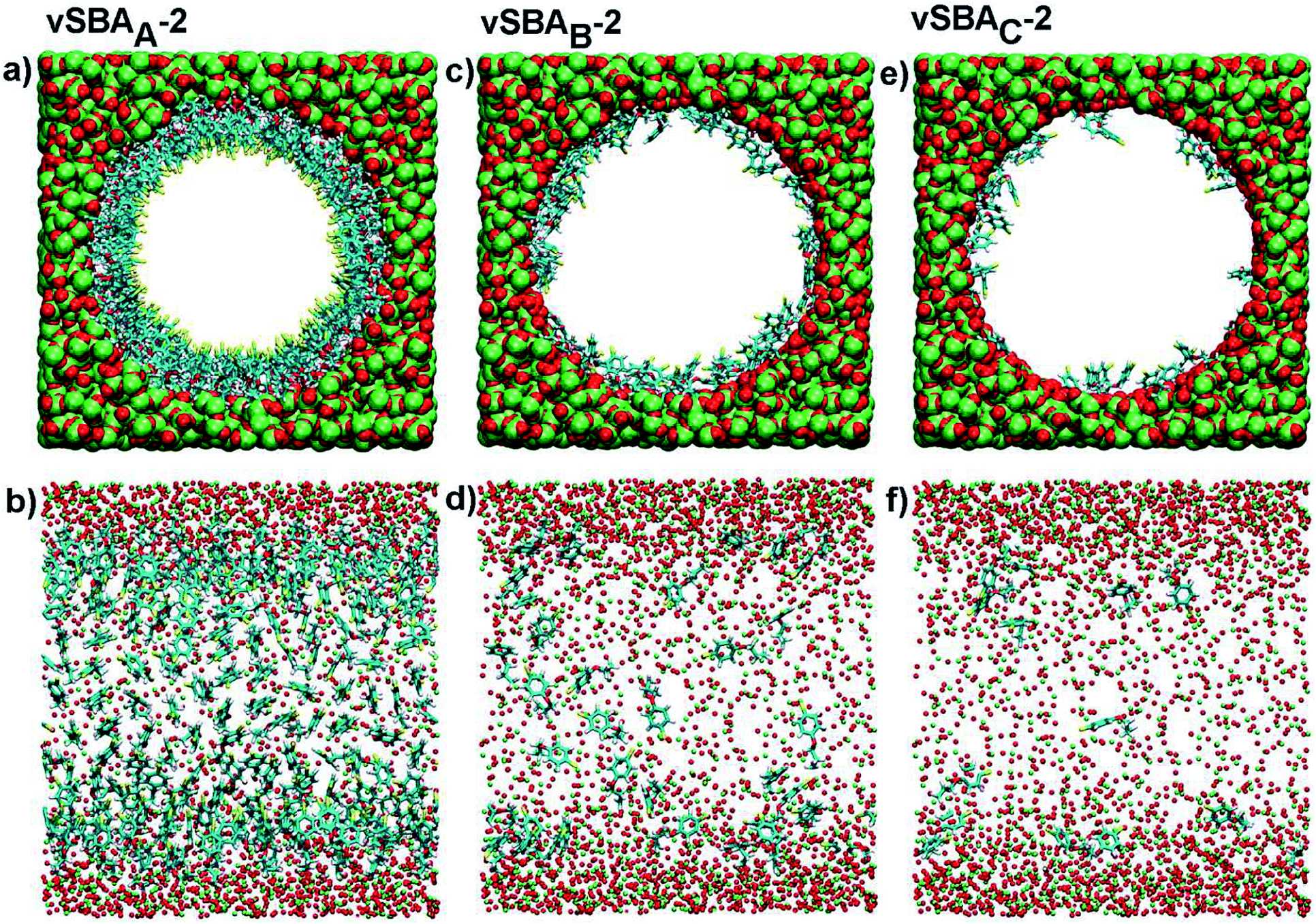 | ||
| Fig. 3 Snapshots of virtual silica pores calculated with DREIDING FF: transverse section (a, c and e) and longitudinal section (b, d and f). Color key: S (yellow), C (blue), H (white), O (red), Si (green). | ||
A striking difference was noticed between the three systems (Fig. 3b, d and f). In the case of a high loading (vSBAA-2), due to the high density of organic moieties, aromatic rings are aligned and sulfur atoms are directed towards the center of the pore.
Conversely, with lower organic loadings, vSBAB-2 and vSBAC-2, the behavior of organic fragments changes completely as they frequently refold over the surface. The inter-chain interactions decrease and the interactions with the surface of the silica increase.
Statistics can be extracted from these simulations. Regardless of the force field, DREIDING or GAFF, the sulfur atoms are, as expected, closer to each other in more highly loaded materials. The curves have the same profile with both force fields. For the sake of simplification, only the DREIDING curves are presented (Fig. 4a).
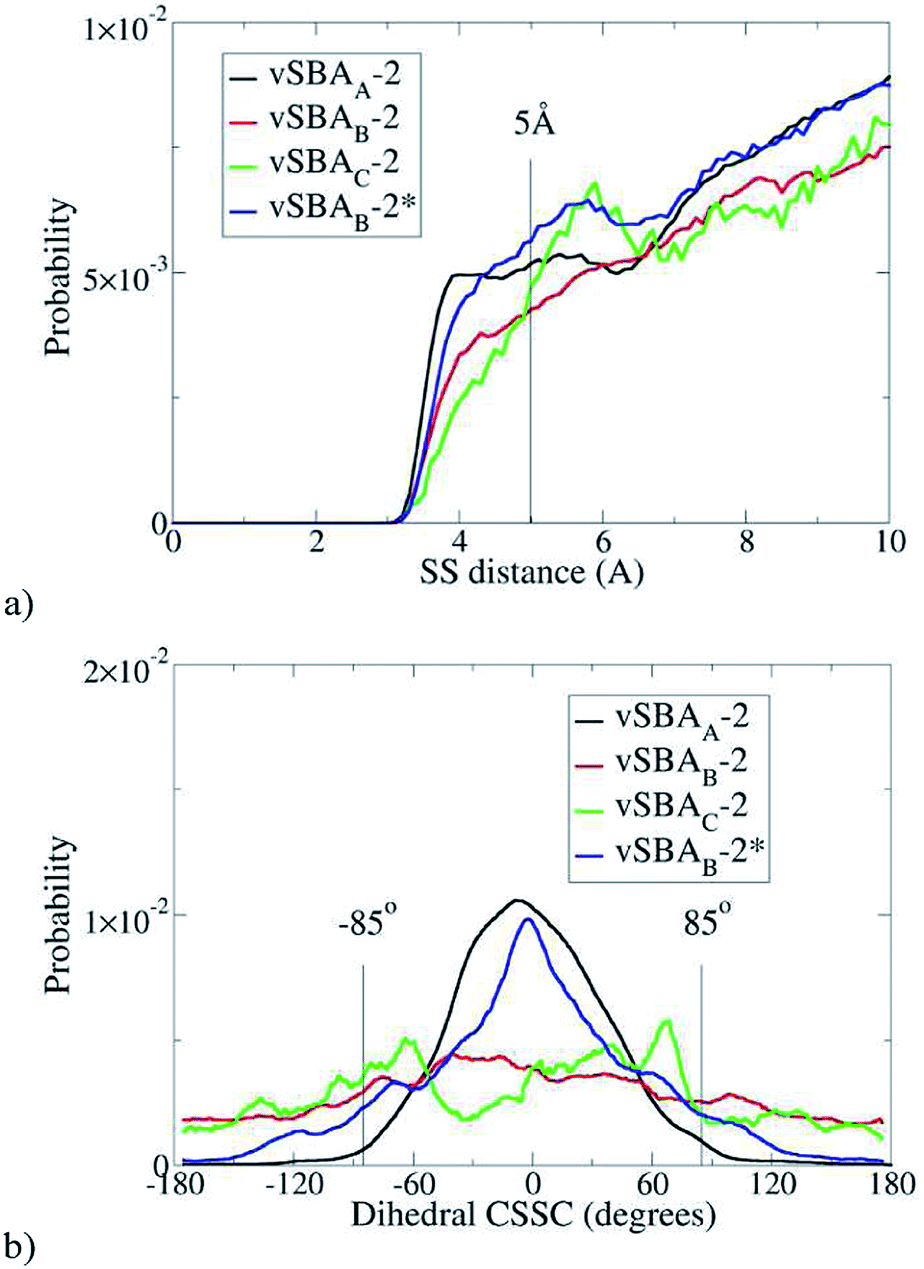 | ||
| Fig. 4 Distance between sulfur atoms (a) and (CSSC) dihedral angle with d(S–S) < 5 Å (b), estimated with DREIDING FF. | ||
As already mentioned, the formation of a disulfide bond can only occur if the two sulfur atoms are reasonably close together. Therefore in the following discussion, only the cases where the inter-sulfur distance is less than 5 Å are considered. The second parameter of interest is the dihedral angle (CSSC) (Fig. 4b).
With vSBAB-2 and vSBAC-2, the dihedral angle can reach a wide range of values; there is no constraint to dimerization. The situation is clearly different for vSBAA-2. Most of the organic moieties have a CSSC dihedral angle close to 0°, which is consistent with aligned aromatic rings directed towards the central axis of the cylinder. This average value does not fit the skew conformation, where the dihedral angle is close to 85°, which is known to be the most stable both experimentally41–43 and theoretically.36,49–51 Any disulfide linkage formed in such a compressed cis conformation would be highly energetic, as stated above. In other words, under this monolayer-like structure the formation of disulfide is hindered. The chain flexibility is locked by steric effects with little influence of the sulfur end group. This kind of packing would similarly occur with thiols instead of sulfanyl radicals at the end of the anchored chains. Therefore, the lack of spin-coupling interactions in the force fields tested is not harmful for this analysis.
These calculations fit quite well the variation of the lifetime recorded for the radicals resulting from SBAn-1 and SBAn-2. The lifetime decreases with the loading down to a minimum, and then increases at high dilution in organic precursors (Table 1). In the case of SBA17-1 and SBA21-2, the precursors form a compact layer on the surface of the pore. With such a spatial arrangement, the formation of the S–S bond in a stable conformation is made difficult because the stereoelectronic factors are unfavorable. As a consequence, an impressive half-life is observed. At higher dilution, the chain length and the inter-chain distance (Table 1, entries b and f) become compatible with the coupling of two arylsulfanyl radicals. The spacing between the organic chains increases, which favors their mobility and facilitates the formation of disulfide bridges as stable conformers. It must be stressed that at very high dilution (SBA239-1), the organic moieties are statistically isolated; they cannot interact with each other, and this results in an increase of t1/2. The formation of the disulfide bond is unlikely. Unidentified reactions with the surface occurred as complex decay curves are observed.
The effects of passivation were also investigated by molecular dynamics. vSBAB-2 was virtually passivated, leading to vSBAB-2*. Because of the similarity between the results obtained from the previous simulations with two different force fields, only the DREIDING force field was used in this case. These calculations show that passivation precludes the refolding of the organic chain on the surface of the pore (Fig. 5).
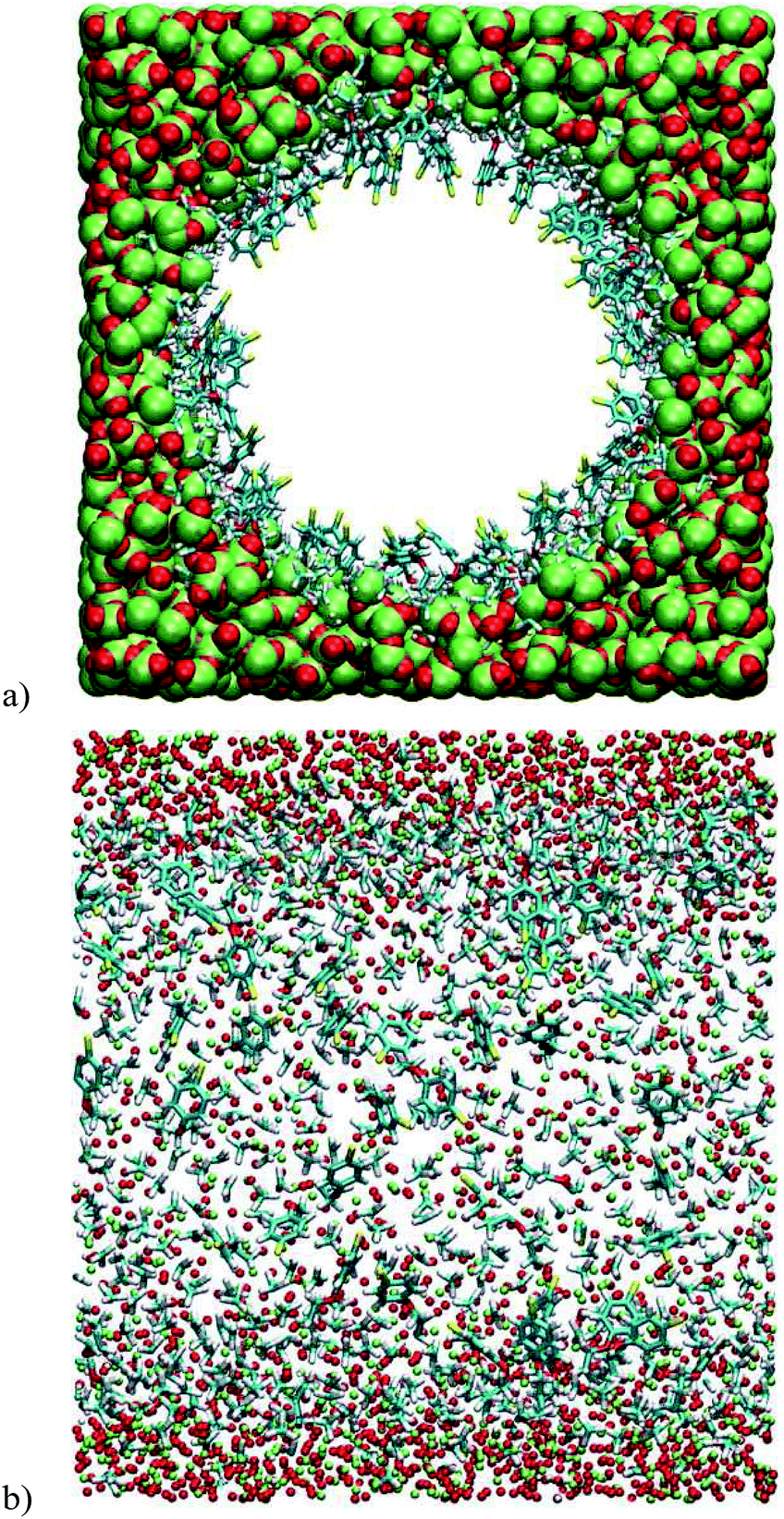 | ||
| Fig. 5 Snapshots of the vSBAB-2* pore calculated with DREIDING FF: (a) transverse section; (b) Longitudinal section. Color key: S (yellow), C (blue), H (white), O (red) and Si (green). | ||
Owing to the steric hindrance of the trimethylsilyl groups, the chains that are found refolded in vSBAB-2 are forced to align in vSBAB-2* with the sulfur atom pointing toward the center of the pore.
Statistics show that the behavior of vSBAB-2* becomes close to that of vSBAA-2 (Fig. 4). The blue line depicting the distribution of S–S inter-distances for vSBAB-2* can almost be superimposed on that of vSBAA-2 for distances of less than 5 Å. With regards to the CSSC dihedral angle, the effect of passivation is very important since the distribution of the values is now centered on 0°; the profile is the same as in the case of vSBAA-2.
The passivation has a direct impact on the structure of the organic monolayer whatever the loading. At high loadings, such as in SBA17-1* and SBA21-2*, the introduction of a trimethylsilyl group on the residual silanol groups reduces the mobility of the organic chains; as a consequence, higher t1/2 values are recorded. With intermediate loadings, as is the case for SBA33-1* and SBA37-2*, the improvement of the half-life is even more pronounced. This can again be rationalized by a lower mobility of the chains parted by the presence of trimethylsilyl groups. The bulky trimethylsilyl groups anchored around the organic chains reduce the length of the flexible part of the organic chains, thus with shorter flexible chains the dimerization is disfavored. The same rationale applies to lower loadings of organic groups, such as in SBA71-1* and SBA73-2*.
The structure of the organic monolayer must have an impact on the shape of the EPR spectra. The line width is a good indicator to probe the radical environment, as it is influenced by the mobility of the radical and its interactions with other radicals.
Line width values and their variations between the beginning and the end of the irradiation are gathered in Table 3 for SBAn-2 and SBAn-2* (see ESI† for SBAn-1 and SBAn-1*).
| Entry | Line width | ΔHpp | ||
|---|---|---|---|---|
| H pp1 (G) | H pp2 (G) | |||
| a Line width at the beginning of the irradiation. b Line width at the end of the irradiation (±0.3 G). | ||||
| a | SBA21-2 | 10.91 | 12.70 | 1.79 |
| b | SBA37-2 | 12.92 | 12.34 | −0.58 |
| c | SBA73-2 | 11.96 | 11.67 | −0.29 |
| d | SBA21-2* | 8.81 | 9.06 | 0.25 |
| e | SBA37-2* | 9.61 | 14.36 | 4.75 |
| f | SBA73-2* | 9.84 | 14.34 | 4.50 |
Line width broadening is the result of three main effects acting together: chemical exchange (conformational changes, tumbling), spin–spin exchange, and dipole–dipole interaction.52 In the case of the low loadings (Table 3, entries b, c vs. entries e, f), the line width at the beginning of the irradiation, that is at low radical concentration, is broader for non passivated silicas than for passivated ones. Due to the low radical concentration, spin–spin exchange and dipole–dipole interaction can be disregarded. This observation indicates that, in non-passivated silicas, organic fragments are less mobile due to their refolding on the surface. This experimental evidence highlights the passivation effect, which prevents the interactions between the silanol groups and the organic chains and thus the refolding on the surface. The line width increases regularly during the irradiation when the silicas are passivated as the concentration of sulfanyl radicals increases due to radical interactions.
For the non-passivated series, no significant increases were observed for SBA37-2 and SBA73-2 meaning that the interactions between the surface and the sulfanyl radicals (chemical exchange) predominated over the spin–spin exchange and dipole–dipole interactions.
For SBA21-2*, the line width is nearly the same at the end of irradiation as at the beginning, and it increases after 80 h (32% decay, Hpp = 13.32 G) (Fig. 6). The initial single symmetrical line (a), observed for strong coupling, evolved towards a broader anisotropic signal (b) with weaker exchange interactions.
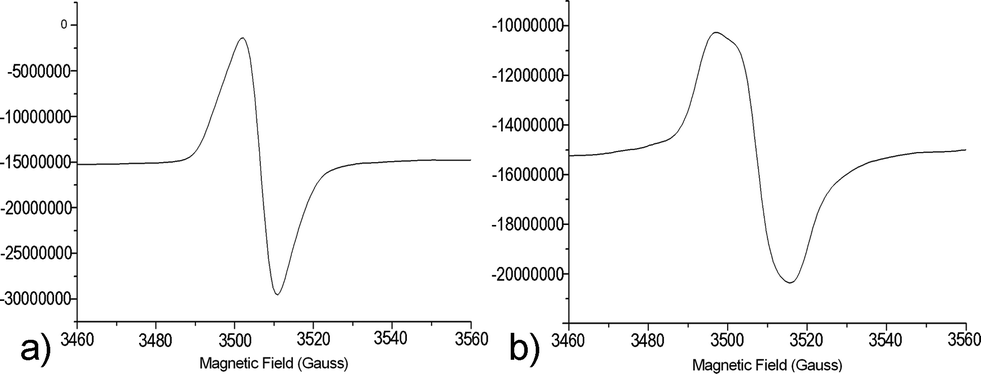 | ||
| Fig. 6 EPR spectra at 293 K of SBA21-2* at the end of the irradiation (Hpp = 9.06 G) (a) and after 80 h (Hpp = 13.32 G) (b). | ||
It is generally accepted that the line width tends to increase linearly with the concentration, due to dipolar interactions. For large enough concentrations, exchange effects become appreciable, and at high concentrations the exchange interaction causes the line to narrow.53Fig. 6 shows the evolution of the signal shape from the highest concentration (a), where spin exchange is predominant, to a lower concentration, where dipolar interactions take over.
The density of radicals in SBA21-2 is large enough for spin–spin exchange or dipole–dipole interaction to occur and induce line broadening.
Conclusions
Room temperature photolysis of covalently linked aromatic thiols, regularly spread over the pores of nanostructured silicas, led to arylsulfanyl radicals with half-lives which can be more than 9 orders of magnitude higher than the lifetime of phenylsulfanyl radical in solution taken as a reference. Counter-intuitively, the higher the density of organic chains on the surface of the pore, the longer the lifetime. Molecular dynamics simulations showed that this huge increase is not only related to the effect of anchoring but also to the monolayer structure of the organic precursors. At high loadings, supramolecular interactions prevent the signal decay, as the dimerization of sulfanyl radicals would lead to unstable conformers of the expected disulfides with mechanical stress disfavoring the process. The passivation of residual silanols on the surface of the pore accentuates the slowing down of the radical decay. The radical centers are spaced apart due to the restricted freedom of motion of the anchoring chains. Increasing the persistence of a radical classified as transient to such a level opens up routes to paramagnetic materials unexplored to date. This versatile approach using confinement effects is likely to be suitable for extension to other types of transient radical species.Acknowledgements
The authors thank Dr P. Llewellyn for critical reading of the manuscript. The authors acknowledge the Agence Nationale de la Recherche for funding (ANR-12-JS07-0005). This work was supported by the computing facilities of the CRCMM, Centre Régional de Compétences en Modélisation Moléculaire de Marseille.Notes and references
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13 CrossRef CAS.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 193 CrossRef CAS.
- M. Newcomb, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, 2012, vol. 1, ch. 5, pp. 107–124 Search PubMed.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321 CAS.
- M. J. Perkins, Adv. Phys. Org. Chem., 1980, 17, 1 CrossRef CAS.
- D. Rehorek, Chem. Soc. Rev., 1991, 20, 341 RSC.
- A. L. J. Beckwith, V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4983 CrossRef CAS.
- V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4992 CrossRef CAS.
- Stable Radicals, ed. R. G. Hicks, Wiley, 2010 Search PubMed.
- J. L. Zweier and P. Kuppusamy, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5703 CrossRef CAS.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303 RSC.
- N. Domingo, E. Bellido and D. Ruiz-Molina, Chem. Soc. Rev., 2012, 41, 258 RSC.
- M. Mas-Torrent, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2012, 112, 2506 CrossRef CAS PubMed.
- N. T. Kartel, Theor. Exp. Chem., 1980, 16, 128 CrossRef CAS and references therein.
- N. J. Turro, Acc. Chem. Res., 2000, 33, 637 CrossRef CAS PubMed.
- N. J. Turro, X.-G. Lei, S. Jockusch, L. Wei, Z. Liu, L. Abrams and M. F. Ottaviani, J. Org. Chem., 2002, 67, 2606 CrossRef CAS PubMed.
- N. J. Turro and S. Jockusch, J. Org. Chem., 2002, 67, 5779 CrossRef CAS PubMed.
- A. Moscatelli, Z. Liu, X. Lei, J. Dyer, L. Abrams, M. F. Ottaviani and N. J. Turro, J. Am. Chem. Soc., 2008, 130, 11344 CrossRef CAS PubMed.
- J. P. Da Dilva, S. Jockusch, J. M. G. Martinho, M. F. Ottaviani and N. J. Turro, Org. Lett., 2012, 12, 3062 CrossRef PubMed.
- S. Panagiota, M. Louloudi and Y. Deligiannakis, Chem. Phys. Lett., 2009, 472, 85 CrossRef CAS PubMed.
- M. K. Kidder, P. F. Britt, Z. Zhang, S. Dai, E. W. Hagaman and A. C. Buchanan, III, J. Am. Chem. Soc., 2005, 127, 6353 CrossRef CAS PubMed.
- M. K. Kidder, P. F. Britt, A. L. Chaffee and A. C. Buchanan, III, Chem. Commun., 2007, 52 RSC.
- M. K. Kidder, A. L. Chaffee, M.-H. T. Nguyen and A. C. Buchanan, III, J. Org. Chem., 2011, 76, 6014 CrossRef CAS PubMed.
- C. T. Kresge and W. J. Roth, Chem. Soc. Rev., 2013, 42, 3663 RSC.
- M. P. Bertrand and C. Ferreri, in Radical in Organic Synthesis, ed. P. Renaud and M. Sibi, Wiley-VCH, Weinheim, 2001, vol. 2, pp. 485–503 Search PubMed.
- C. Chatgilialoglu, M. P. Bertrand and C. Ferreri, in S-Centered Radicals, ed. Z. B. Alfassi, Wiley, Chichester, 1999, pp. 311–354 Search PubMed.
- F. Deéneès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587 CrossRef PubMed.
- T. W. Scott and S. N. Liu, J. Phys. Chem., 1989, 93, 1393 CrossRef CAS.
- K.-D. Asmus and M. Bonifacic, in S-Centered Radicals, ed. Z. B. Alfassi, Wiley, Chichester, 1999, pp. 141–191 Search PubMed.
- Y. V. Razskazovskii and M. Y. Mel'nikov, in S-Centered Radicals, ed. Z. B. Alfassi, Wiley, Chichester, 1999, pp. 225–244 Search PubMed.
- F. C. Thyrion, J. Phys. Chem., 1973, 77, 1478 CrossRef CAS.
- J. C. Walton, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, 2012, vol. 1, ch. 7, pp. 151–174 Search PubMed.
- U. Schmidt, Angew. Chem., Int. Ed., 1964, 3, 602 CrossRef PubMed.
- W. Rundel and K. Scheffler, Angew. Chem., Int. Ed., 1965, 4, 243 CrossRef PubMed.
- U. Schmidt and A. Müller, Angew. Chem., Int. Ed., 1963, 2, 216 CrossRef PubMed.
- G. N. R. Tripathi, Q. Sun, D. A. Armstrong, D. M. Chipman and R. H. Schuler, J. Phys. Chem., 1992, 96, 5344 CrossRef CAS.
- F. Gerson and W. Huber in Electron Spin Resonance Spectroscopy of Organic Radicals, ed. F. Gerson and W. Huber, Wiley-VCH, 2003, pp. 97–165 Search PubMed.
- N. A. Borisevich, V. I. Buganov, V. L. Dobovskii, S. A. Tikhomirov and G. B. Tolstorozhev, Opt. Spectrosc., 2005, 98, 368 CrossRef CAS.
- A. G. Vandeputte, M.-F. Reyniers and G. B. Marin, J. Phys. Chem. A, 2010, 114, 10531 CrossRef CAS PubMed.
- (a) R. Benassi, G. L. Fiandri and F. A. Taddei, J. Mol. Struct., 1997, 418, l27 Search PubMed; (b) L.-F. Zou, K. Shen, Y. Fu and Q.-X. Guo, J. Phys. Org. Chem., 2007, 20, 754 CrossRef CAS PubMed.
- M. Sacerdoti and G. Gilli, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 327 CrossRef.
- T. Shimizu, H. Isonon, M. Yasui, F. Iwasaki and N. Kamigata, Org. Lett., 2001, 3, 3639 CrossRef CAS PubMed.
- T. Schaefer, S. R. Salman, T. A. Wildman and P. D. Clark, Can. J. Chem., 1982, 60, 342 CrossRef.
- I. B. Baldus and F. Gräter, Biophys. J., 2012, 102, 622 CrossRef CAS PubMed.
- N. L. Haworth, J. Y. Liu, S. W. Fan, J. E. Gready and M. A. Wouters, Aust. J. Chem., 2010, 63, 379 CrossRef CAS.
- E. Dumont, J.-F. Loos and X. Assfeld, Chem. Phys. Lett., 2008, 458, 276 CrossRef CAS PubMed.
- M. F. Iozzi, T. Helgaker and E. Uggerud, J. Phys. Chem. A, 2011, 115, 2308 CrossRef CAS PubMed.
- A. Brodka and T. W. Zerda, J. Chem. Phys., 1996, 104, 6319 CrossRef CAS PubMed.
- D. Nori-Shargh, H. Yahyaei and J. E. Boggs, J. Mol. Graphics Modell., 2010, 28, 807 CrossRef CAS PubMed.
- R. S. Sengar, V. N. Nemykin and P. Basu, New J. Chem., 2003, 27, 1115 RSC.
- N. L. Haworth, J. Y. Liu, S. W. Fan, J. E. Gready and M. A. Wouters, Aust. J. Chem., 2010, 63, 379 CrossRef CAS.
- (a) Electron Paramagnetic Resonance, ed. J. A. Weil and J. R. Bolton, John Wiley & Sons, 2007 Search PubMed; (b) Principles of Electron Spin Resonance, ed. N. M. Atherton, Ellis Horwood, 1993 Search PubMed.
- K. M. Salikhov, Appl. Magn. Reson., 2010, 38, 237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization for the organic compounds and the derived materials (NMR, TEM, SAXS, nitrogen adsorption/desorption analysis, ATG). EPR decay curves, variation of EPR line width for SBAn-1 and SBAn-1*, spin density calculations, DFT calculations and molecular dynamics simulations. See DOI: 10.1039/c4sc01907e |
| This journal is © The Royal Society of Chemistry 2014 |