DOI:
10.1039/C4SC01432D
(Edge Article)
Chem. Sci., 2014,
5, 3627-3633
Quinoidal diindenothienoacenes: synthesis and properties of new functional organic materials†‡
Received
15th May 2014
, Accepted 16th June 2014
First published on 16th June 2014
Abstract
We report the preparation and characterization of a new class of quinoidal thienoacenes. The synthetic route is efficient, high-yielding and scalable with the potential for further functionalization. Single crystal X-ray diffraction reveals that, as size increases, the molecules pack in progressively closer 1D arrangements. The title compounds are shown to have amphoteric redox behaviour by cyclic voltammetry. The anion radicals are studied by EPR spectrometry and by computations. The electron-accepting nature, NIR absorption and the low-lying LUMO energies (ca. −4.0 eV) allude to potential use in materials applications.
Introduction
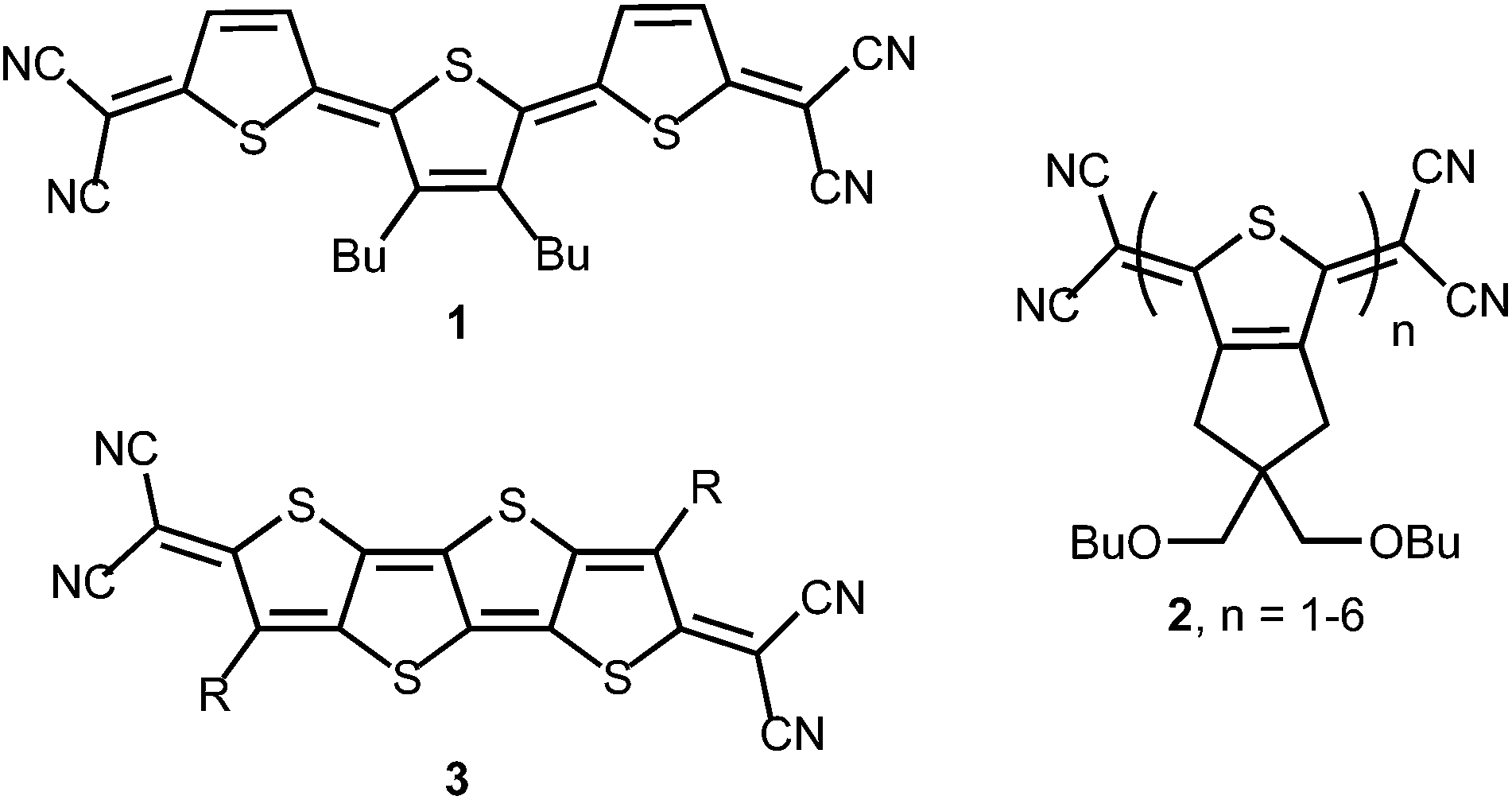
Quinoidal oligothiophenes (QOTs) and quinoidal thienoacenes (QTAs) have attracted significant interest as functional organic materials (Fig. 1). Initially, the preparation of thiophene oligomers and study of their reduced and oxidized forms provided models for the doped portions of polythiophene.1 One of the first small molecules to exhibit ambipolar character in an OFET was based on a terthienoquinoidal core (1).2 Thorough understanding of the stability of the reduced and oxidized states of 1 led to increased OFET performance and the discovery of ambipolar transport characteristics.3 Otsubo, Aso and coworkers accomplished the synthesis of the largest family of QOTs (2).4 Solubility issues of the larger QOTs were solved by fusion of a solubilizing bis(butoxymethyl)cyclopentane unit to the thiophene core. The longest members (n = 5, 6) featured open-shell ground states and low-energy absorptions to 1400 nm.5 A terthiophene derivative of 2 as the active layer in OFETs was also described.6 Recently, Zhu et al. reported n-channel behaviour in thin films of QTA 3.7 As noted by the authors, isomerization issues inherent in QOTs are eliminated by moving to the fused QTA core. Lastly, the QTA scaffolds are promising for nonlinear optics due to their rigidity and symmetry.8 Large alternant polycyclic hydrocarbons possessing low energy triplet excited states and multiphoton absorption properties are of interest for singlet exciton fission.9
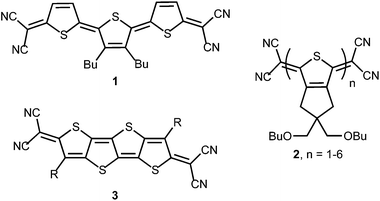 |
| | Fig. 1 Examples of quinoidal oligothiophenes (1 and 2) and thienoacenes (3) reported in the literature; R = solubilizing alkyl group. | |
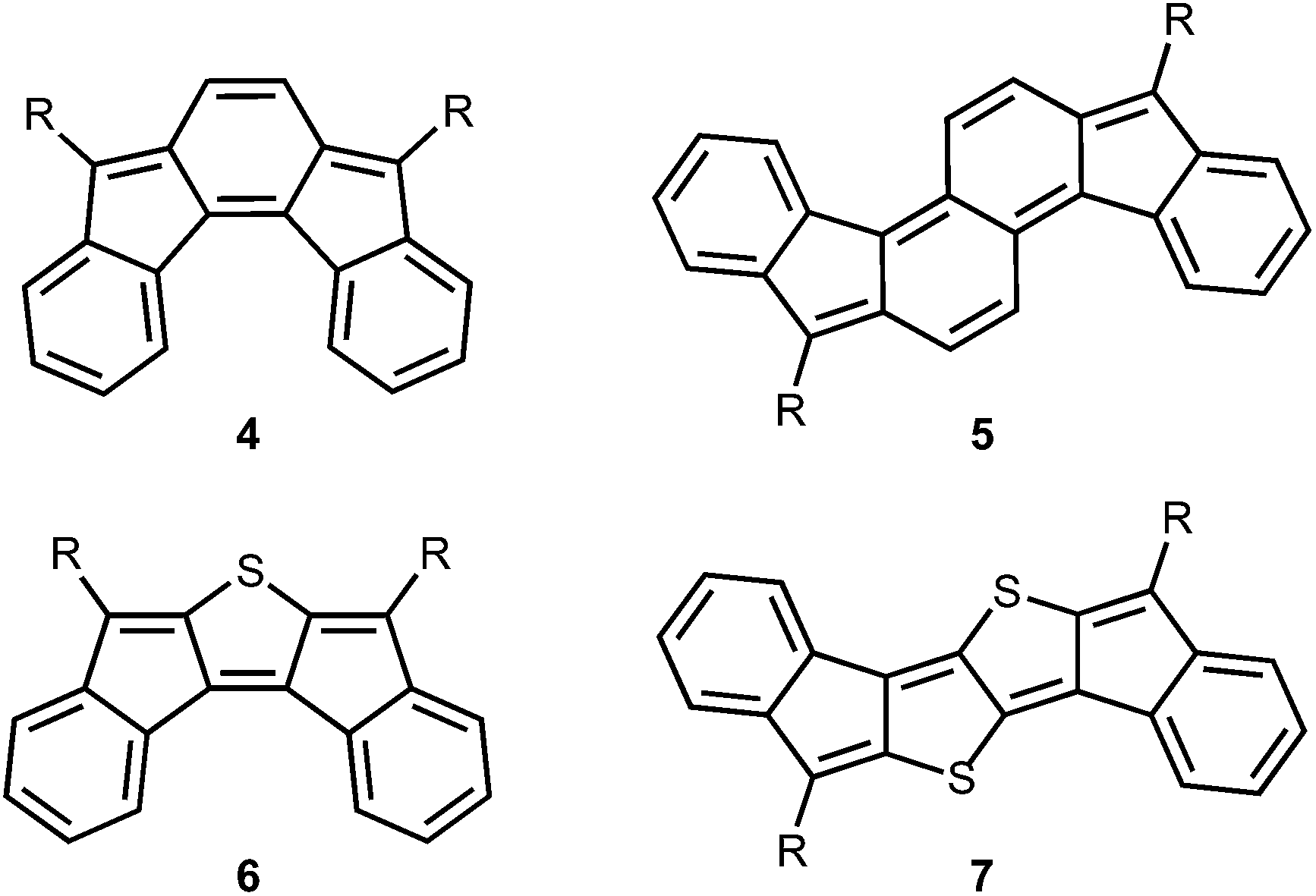
Our group has initiated studies on a seldom explored class of quinoidal molecules based on the indenofluorene skeleton.10 While most of our work has focused on indeno[1,2-b]fluorenes,11 we have reported derivatives of indeno[2,1-c]fluorene 412 and fluoreno[4,3-c]fluorene 5 (Fig. 2),13 which exhibit interesting absorption profiles and amphoteric redox behaviour. Very recently we reported the fully conjugated indacenedithiophenes,14 where thiophenes replaced the outer benzene rings of indeno[1,2-b]fluorenes. In analogy, exchange of the bridging sp2 carbon units in 4 and 5 with isoelectronic sulfur atoms to provide 6 and 7 is an attractive and straightforward approach to further tune the quinoidal core of indenofluorenes. Sulfur incorporation into the framework of polycyclic hydrocarbons is a versatile method to promote good solid-state ordering and improve stability at ambient conditions.15
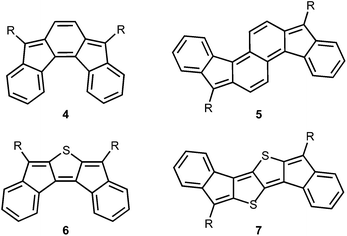 |
| | Fig. 2 Previously reported indenofluorene 4 and fluorenofluorene 5 and their DI[n]T analogues 6 and 7. | |
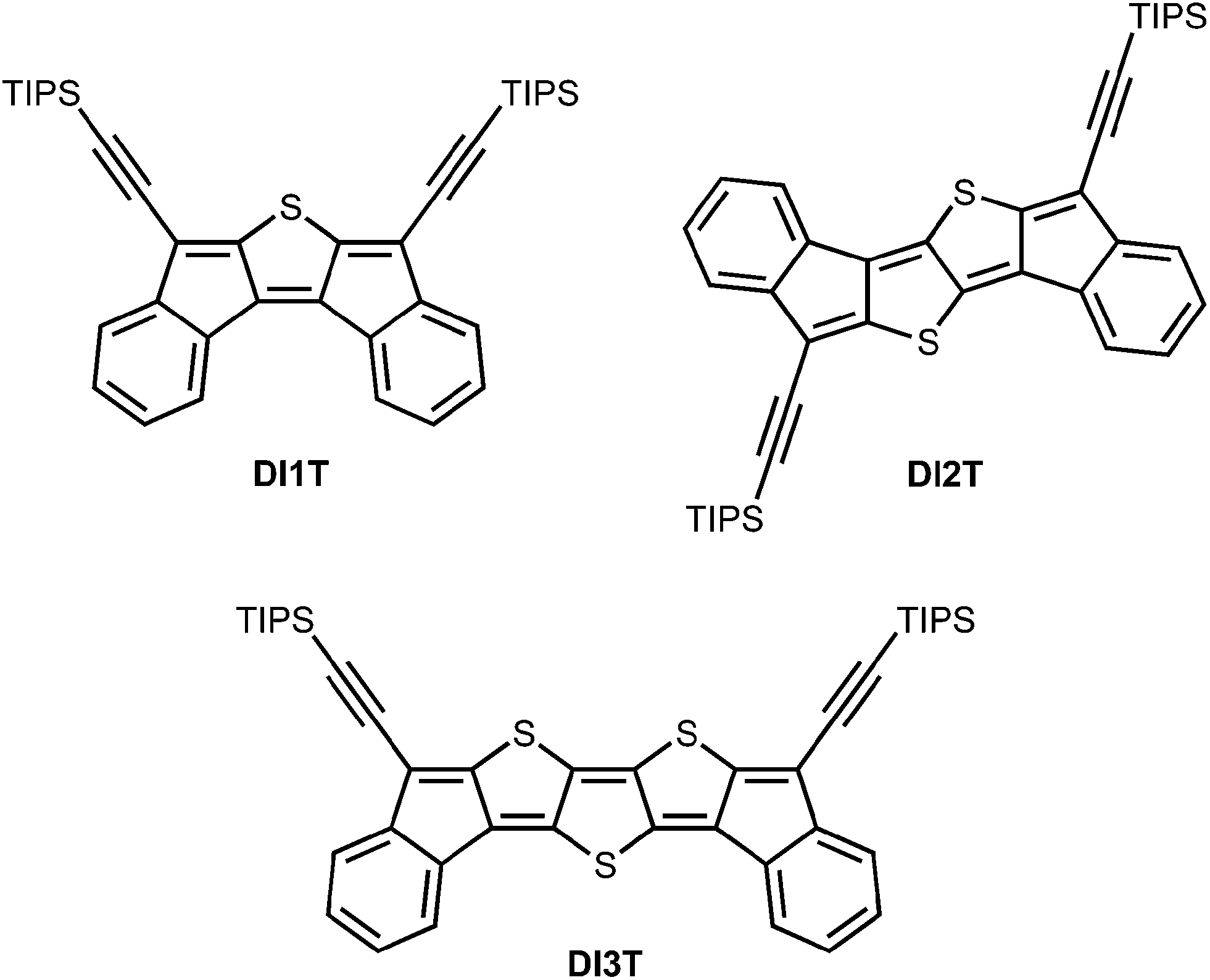
Herein we describe the synthesis and characterization of a series of quinoidal diindeno(thieno)thiophenes (DI[n]Ts) (Fig. 3). This work represents a new approach to quinoidal thienoacenes through the fusion of electron-accepting indene fragments to a thienoacene core. The ease of thiophene synthesis and anticipated stability of the final quinoidal molecules are appealing features of this strategy. Possible substituents include (trialkylsilyl)ethynyl groups to favour solid-state order16 and aryl groups to tune the electronics11b,c of the architecture.
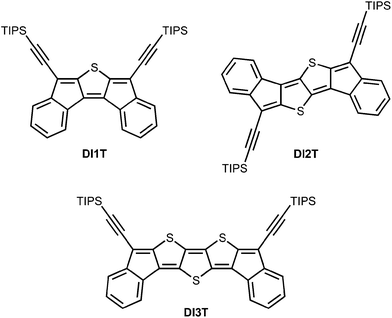 |
| | Fig. 3 Structures of diindeno(thieno)thiophenes (DI[n]T). | |
Results and discussion
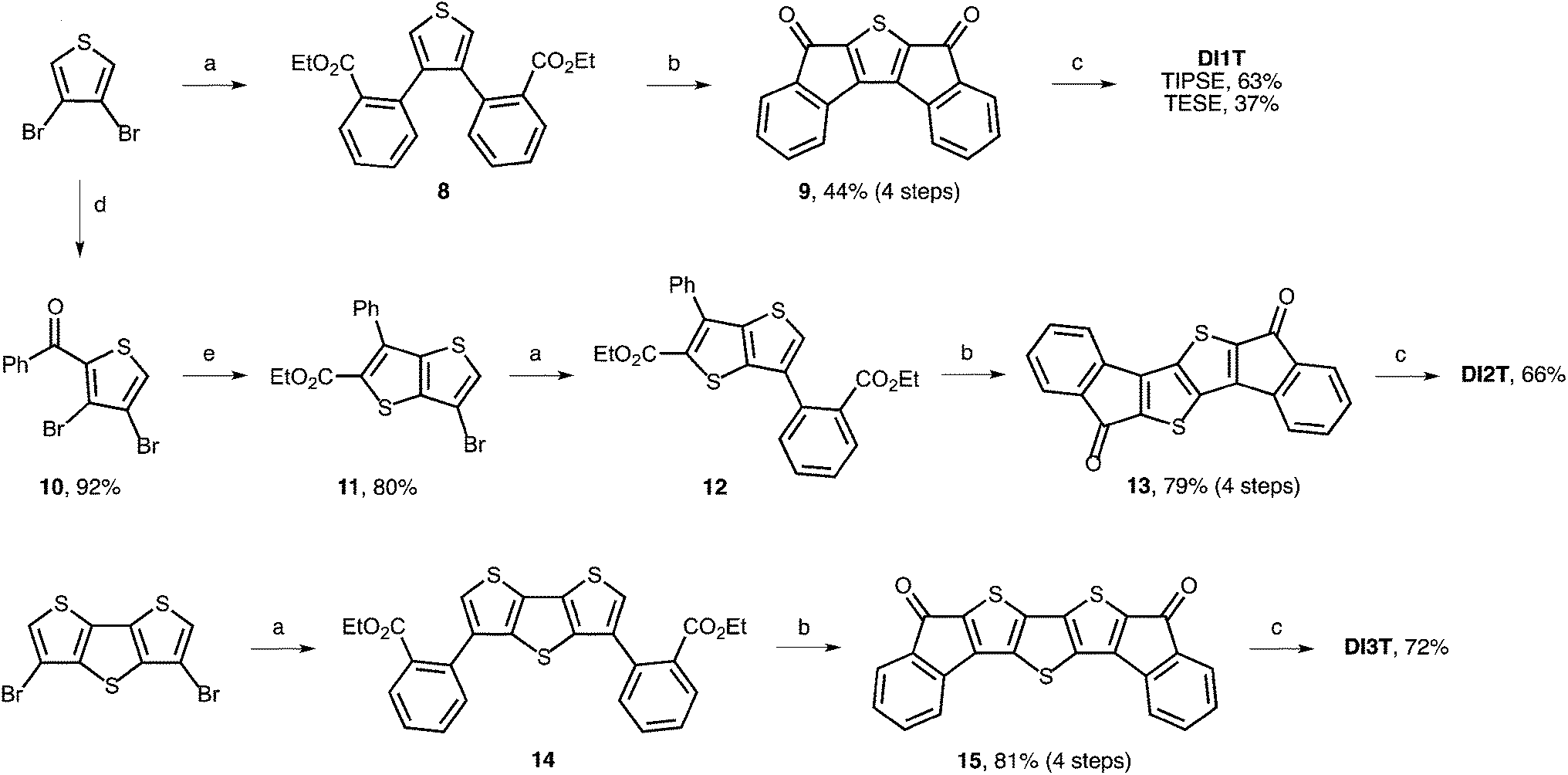
Synthesis is initiated by Suzuki–Miyaura cross-coupling of 3,4-dibromothiophene and commercially available 2-ethoxy-carbonylbenzeneboronic acid (Scheme 1). Use of Buchwald's SPhos ligand was critical for efficient coupling of the electron-rich bromothiophene with the electron-poor arylboronic acid.17 Next, saponification of diester 8 followed by formation of the acid chloride and Friedel–Crafts acylation provides dione 9 in good yield. Gratifyingly, we found that 3,4-dibromothiophene could be mono-acylated to give 10 in 92% yield. Condensation with ethyl thioglycolate in the presence of potassium carbonate furnishes thienothiophene 11 in 80% yield. This route avoids the preparation of unsubstituted thieno[3,2-b]thiophene either through traditional methods18 or the improved Matzger route.19 Suzuki–Miyaura cross-coupling gives diester 12 in quantitative yield. Elaboration to 13 proceeds in 79% yield over four steps.
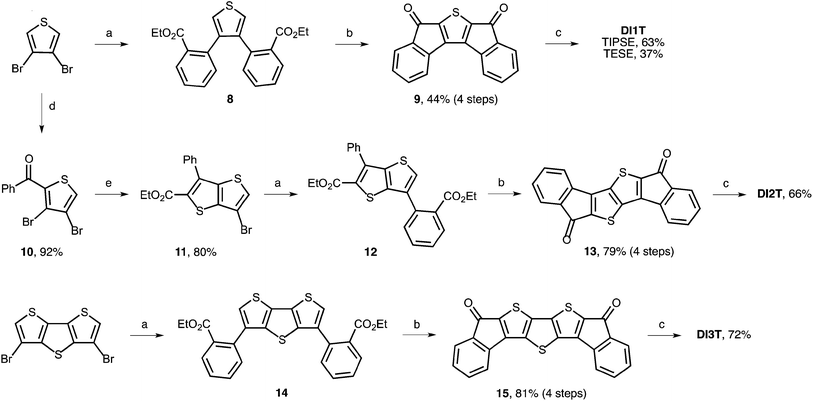 |
| | Scheme 1 Synthesis of DI1T, DI2T and DI3T; reagent and conditions: (a) 2-ethoxycarbonylbenzeneboronic acid, Pd2dba3, SPhos, K3PO4, toluene, 100 °C; (b) (i) KOH, EtOH, reflux; (ii) oxalyl chloride, DMF, CH2Cl2, rt; (iii) AlCl3, CH2Cl2, 0 °C to rt; (c) (i) (trialkylsilyl)ethynyllithium, 0 °C, then sat. NH4Cl; (ii) SnCl2, toluene, rt; (d) benzoyl chloride, AlCl3, 0 °C to rt; (e) ethyl thioglycolate, K2CO3, DMF, 60 °C. TIPSE = (triisopropylsilyl)ethynyl, TESE = (triethylsilyl)ethynyl. | |
Dione 15 was prepared in an analogous manner via diester 14 starting from 3,5-dibromodithieno[3,2-b:2′,3′-d]thiophene. Notably, the synthesis of diones 9, 13 and 15 can be performed on multi-gram scale with no silica gel chromatography (see ESI‡ for full details). Nucleophilic addition of (trialkylsilyl)ethynyl lithium proceeds quantitatively despite poor solubility of the dione starting materials. Reduction of the respective diols by anhydrous SnCl2 in toluene gives the quinoidal DI[n]Ts in modest to very good yield. Interestingly, the reduction to DI3T is complete within minutes while the reduction to DI1T and DI2T requires several hours to reach full conversion. The final compounds were stable toward silica gel and ambient conditions. No measures to protect the compounds from air or water were taken and no significant decomposition was observed in solution or in the solid-state.
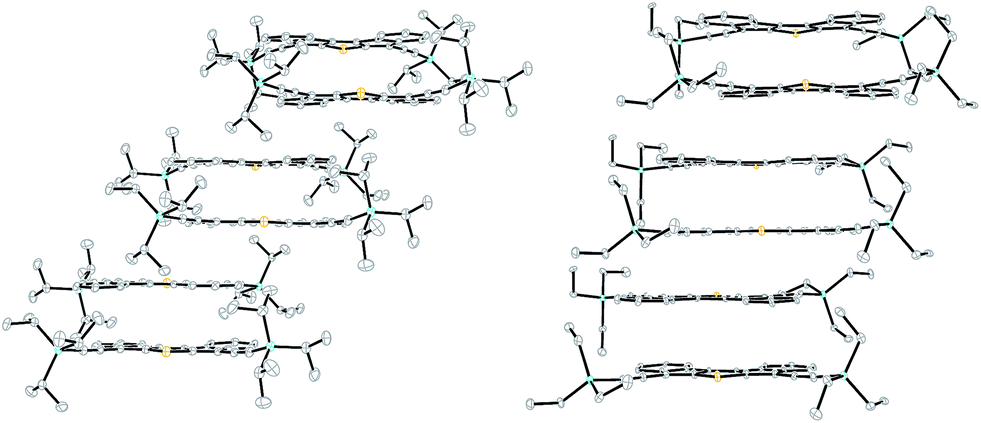
Single crystals of DI[n]Ts sufficient for characterization by X-ray diffraction were obtained by slow evaporation from CH2Cl2. DI1T-TIPSE arranges into a pairwise slipped stacks with distances between average planes of 3.30 Å within the pair and 3.37 Å between adjacent pairs (Fig. 4, left). Lateral (short-axis) slip of the core was found to be 1.06 Å in the pair and 5.82 Å between pairs. DI1T-TESE also packs in pairwise slipped stacks with the distance between average planes of the molecules of 3.43 Å in the pair and 3.34 Å between pairs (Fig. 4, right). The smaller trialkylsilyl group altered the lateral slip to 2.52 Å in the pair and 2.55 Å between pairs. DI2T forms an alternating 1D structure rather than the dimers seen with DI1T due to its centrosymmetry (Fig. 5, top). The distance between the average planes is 3.41 Å with a lateral slip of 1.46 Å between cores of the molecules. Spacing between neighbouring 1D stacks was greater than van der Waals distance. DI3T shows strong overlapping of the quinoidal cores with inter-planar distance of 3.46 Å and lateral slip of 1.79 Å and 1.23 Å (Fig. 5, bottom). The 1D columns are essentially insulated from adjacent columns by the (triisopropylsilyl)ethynyl groups.
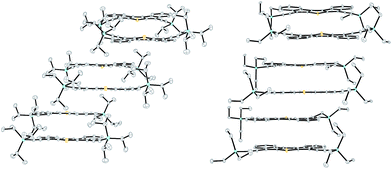 |
| | Fig. 4 Solid-state packing of DI1T-TIPSE (left) and DI1T-TESE (right). | |
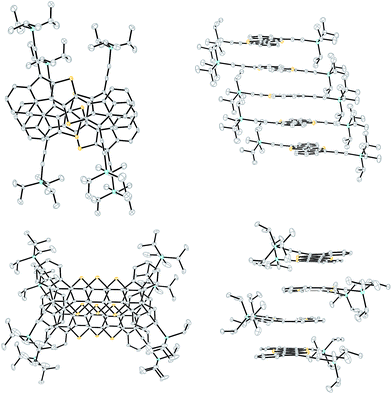 |
| | Fig. 5 Solid-state packing of DI2T (top) and DI3T (bottom). | |
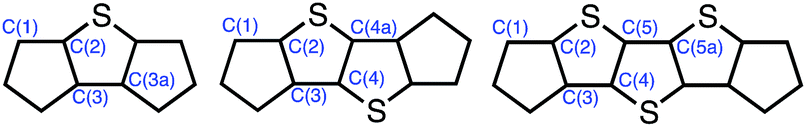
Table 1 summarizes the bond distances in the core of the DI[n]T family.20 All three show distinct bond alternation with the double bonds averaging 1.36–1.37 Å and single bonds 1.44–1.45 Å, fully supporting the proposed quinoidal structures. These values are also in good agreement with those observed in their purely hydrocarbon analogues.12,13 The peripheral benzene rings in the DI[n]Ts have an average bond distance of 1.390 Å with a standard deviation of 0.006 Å. The molecules are essentially planar, as the root-mean-square (RMS) deviation from the average molecular plane is negligible; DI1T-TIPSE showed the greatest RMS deviation of only 0.042 Å.21
Table 1 Solid-state packing and bond distances in the quinoidal corea
|
|
DI1T-TIPSE
|
DI1T-TESE
|
DI2T
|
DI3T
|
All distances in Å; atom numbering is shown below; calculated (UCAM-B3LYP/6-31G(d,p)) bond lengths in brackets.
Values determined omitting the (trialkylsilyl)ethynyl groups.
Average value due to asymmetry in the solved crystal structure or crystallographically independent molecules. 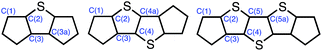
|
| Interplanar distanceb |
3.30, 3.37 |
3.43, 3.34 |
3.41 |
3.46 |
| Lateral slipb |
1.06, 5.82 |
2.52, 2.55 |
1.46 |
1.23, 1.79 |
| RMS deviation from planarityb |
0.042 |
0.026 |
0.013 |
0.026 |
| C(1)–C(2) |
1.365(3) [1.357] |
1.364(12)c |
1.373(5) [1.360] |
1.368(4)c [1.361] |
| C(2)–C(3) |
1.454(2)c [1.456] |
1.454(12)c |
1.447(5) [1.456] |
1.439(4)c [1.455] |
| C(3)–C(3a/4) |
1.359(3) [1.353] |
1.358(11)c |
1.366(5) [1.351] |
1.367(4)c [1.352] |
| C(4)–C(4a/5) |
— |
— |
1.440(7) [1.461] |
1.437(4)c [1.454] |
| C(5)–C(5a) |
— |
— |
— |
1.366(4)c [1.352] |
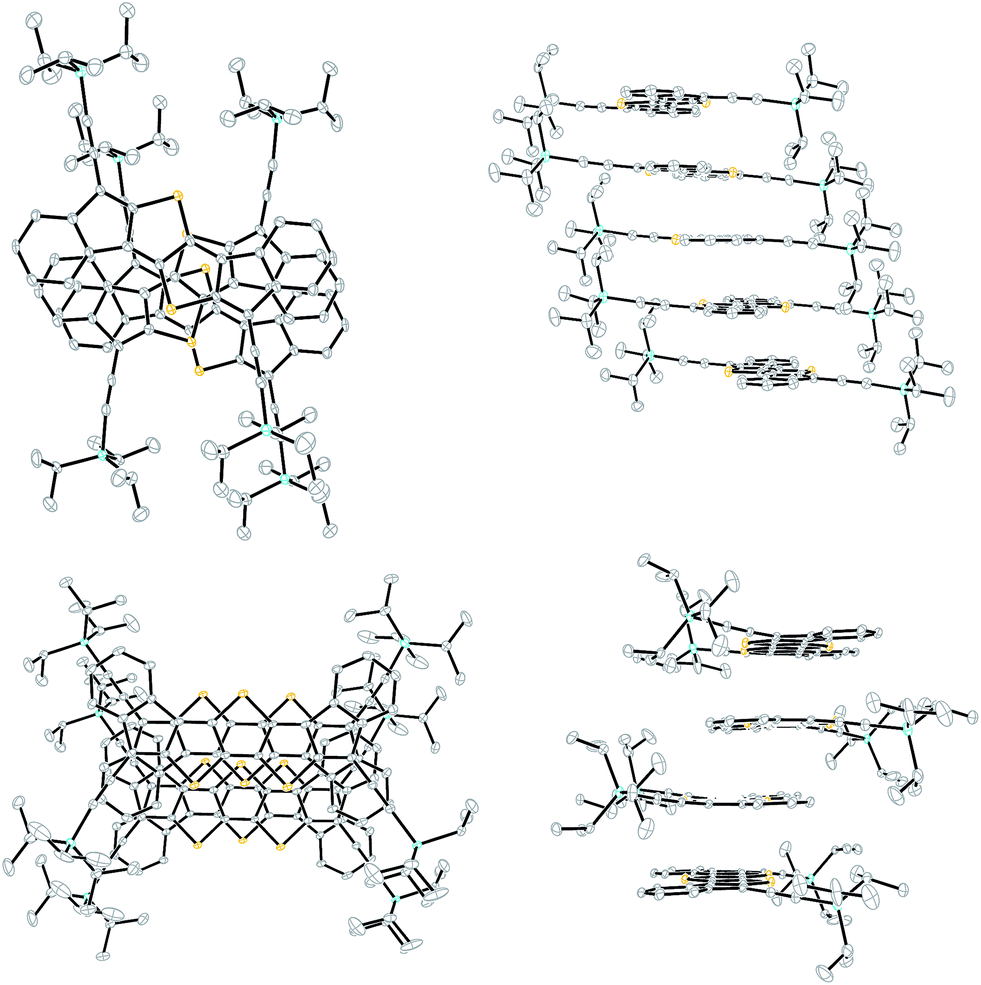
In addition to the DI[n]Ts, we were able to obtain single crystals suitable for X-ray diffraction of diones 9 and 13 from CHCl3/cyclohexane and upon slow cooling from refluxing nitrobenzene, respectively (Fig. 6).22 Dione 9 alternates its orientation within the 1D stack such that the molecular arrangement would result in a net dipole; however, the dipole of the neighbouring stack balances the opposing dipole (see Fig. S4‡). Dione 13 shows a more ordered 1D arrangement due to its centrosymmetry. The distance between the average planes of 9 and 13 are essentially identical at 3.39 Å and 3.38 Å, respectively. Dione 9 has a significant RMS deviation from planarity of 0.079 Å while 13 is nearly planar at 0.027 Å. This is likely due to the steric crowding of the hydrogens in the bay region of 9. Interestingly, the carbonyl oxygens of 9 and 13 show a strong interaction with the bay carbons of adjacent stacks with distances of 3.25 Å for 9 and 3.28 Å for 13.
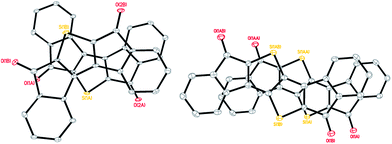 |
| | Fig. 6 Solid-state packing of diones 9 (left) and 13 (right). | |
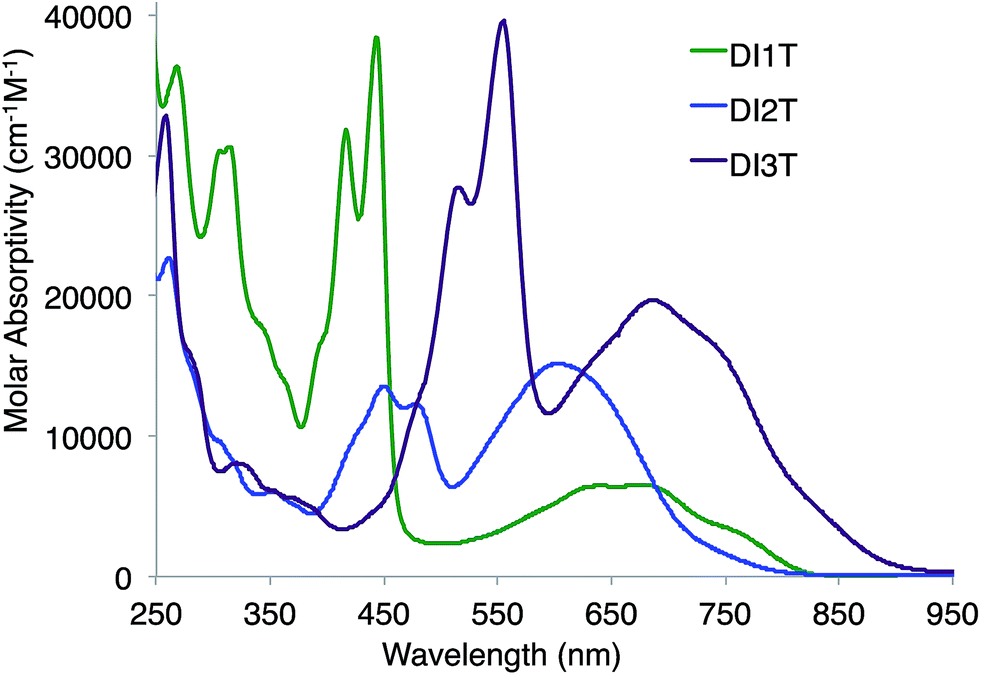
The deeply colored DI[n]Ts were characterized further by absorption spectroscopy (Fig. 7). All compounds exhibit strong, acene-like vibronic features from 350–600 nm and low energy absorptions reaching into the NIR (800–925 nm). The absorption edges are staggered, possibly as a result of the family's alternating axo-/centro-symmetry. Interestingly, DI1T and DI2T exhibit similar absorbance profiles to the related indeno[2,1-c]fluorene and fluoreno[4,3-c]fluorene derivatives (Fig. S1‡). For the DI[n]Ts, the high energy bands red shift by ca. 50–70 nm and the low energy bands blue shift by ca. 25–35 nm. Analogues to 4 and 5, the DI[n]Ts are non-emissive.
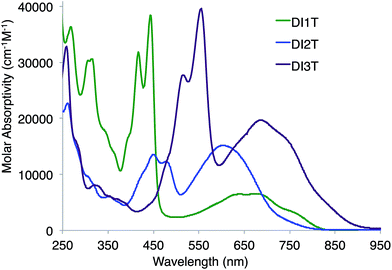 |
| | Fig. 7 Electronic absorption spectra of DI[n]Ts (n = 1–3) in CH2Cl2. | |
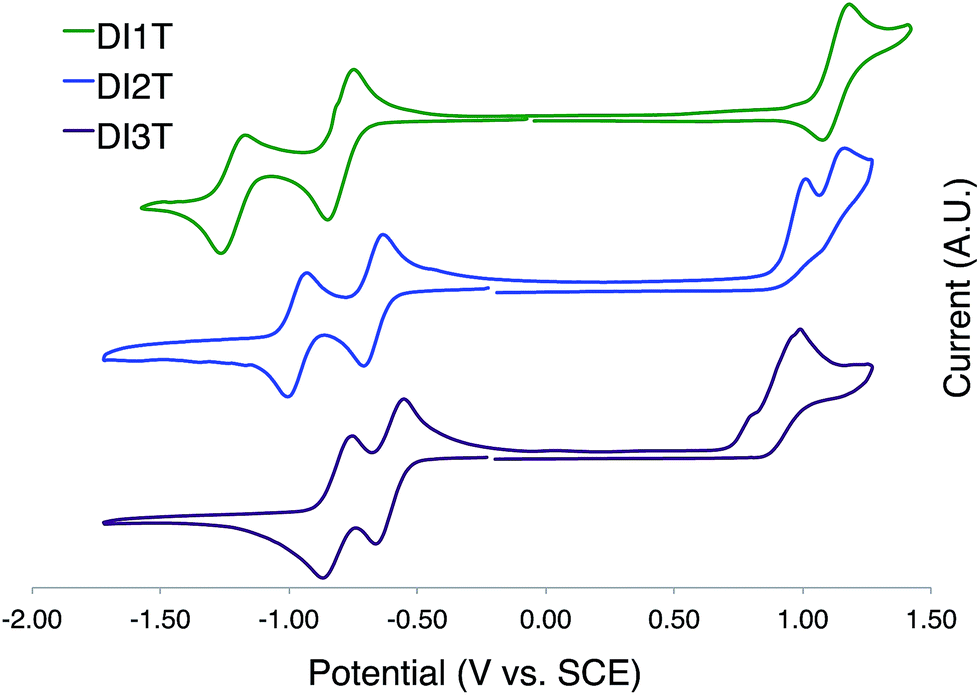
DI[n]Ts exhibit two reversible one electron reductions in solution as examined by cyclic voltammetry (Fig. 8 and Table 2). DI1T shows a reversible oxidation while the oxidation of DI2T and DI3T are essentially irreversible. The difference between E1red and E2red decreases by ca. 0.1 V upon sequential expansion of the quinoidal core. This is most likely due to the mitigation of Coulombic repulsion in the doubly reduced species.24 The values for Eox appear to approach a constant, indicating that incorporation of additional thiophene units does not alter the ionization potential to a large degree. LUMO and HOMO energy levels were derived from the E1red and Eox values, respectively. Egap decreases over the series in nonlinear fashion.
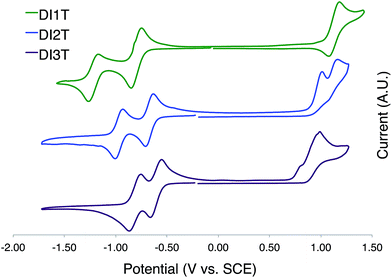 |
| | Fig. 8 Cyclic voltammograms of DI[n]Ts (n = 1–3). | |
Table 2 Electrochemical data for DI[n]Ts (n = 1–3)a
| Compd |
E
1red
|
E
2red
|
E
ox
|
LUMO |
HOMO |
E
gap
|
|
Values reported as the half-wave potential (vs. SCE) using the Fc/Fc+ couple (0.46 V) as an internal standard. HOMO and LUMO energy levels in eV were approximated using SCE = −4.68 eV vs. vacuum (see ref. 23) and E1/2 values for reversible processes or Ep values for irreversible processes; Egap = LUMO − HOMO. Calculated energy levels (B3LYP/6-31G(d,p)) in parentheses.
|
|
DI1T
|
−0.80 |
−1.22 |
1.13 |
−3.84 |
−5.77 |
1.93 |
|
DI2T
|
−0.67 |
−0.97 |
1.02 |
−3.97 |
−5.66 |
1.69 |
|
DI3T
|
−0.61 |
−0.81 |
0.99 |
−4.03 |
−5.63 |
1.60 |
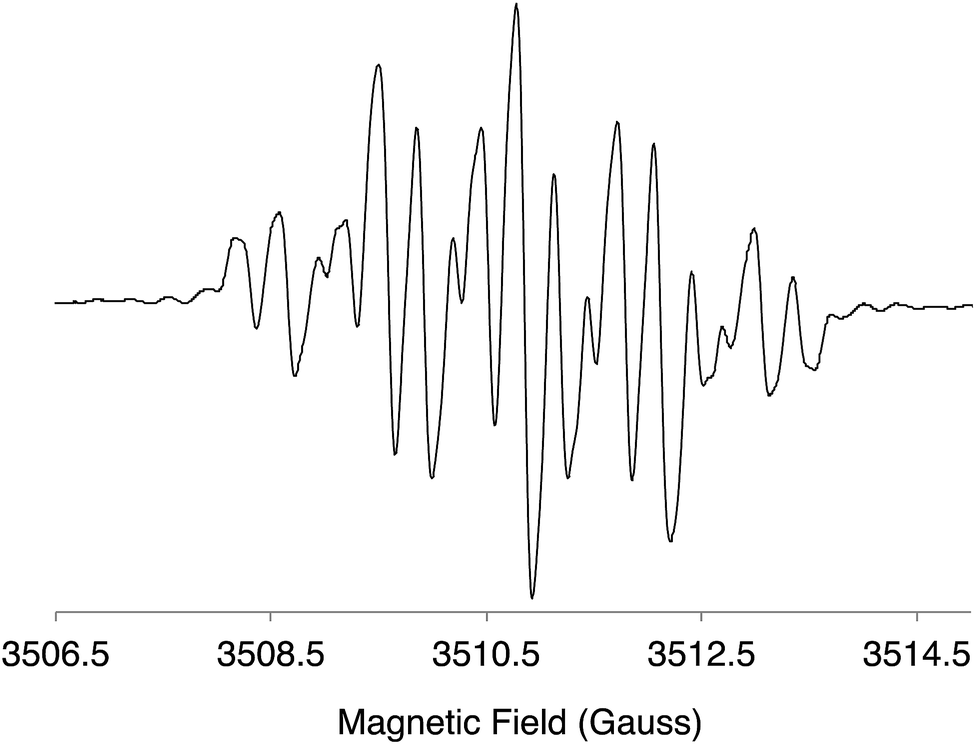
The DI[n]T anion radicals were obtained by reduction of the neutral species with K metal in THF. The EPR spectrum of a blue solution of DI1T˙− is shown in Fig. 9; see ESI for the EPR spectra of DI2T˙− (Fig. S8‡) and DI3T˙− (Fig. S9‡). The hyperfine coupling constants (HFCCs) of the spin active nuclei were determined and experimental carbon spin densities (ρc) were calculated by the McConnell equation (Tables 3 and S1‡). Due to the lack of spin active nuclei on the core of DI[n]T, ρc could not be directly calculated. Very little spin density is contained within the fused benzene rings (0.004–0.044), with more contained in the ethynyl group (0.094–0.112). The approximate spin densities remaining in the core for DI1T, DI2T, and DI3T are 0.59, 0.64, and 0.66, respectively, indicating that a majority of the spin density resides in the thienoacene unit.
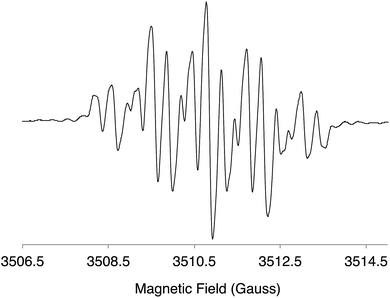 |
| | Fig. 9 EPR spectrum of the radical anion of DI1T. | |
Table 3 Carbon spin densities (ρc)a
| Position |
DI1T
|
DI2T
|
DI3T
|
See ESI for full details; labeling scheme on right. 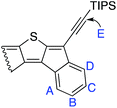
|
| A |
0.034 |
0.022 |
0.024 |
| B |
0.012 |
0.016 |
0.014 |
| C |
0.044 |
0.029 |
0.028 |
| D |
0.004 |
0.010 |
0.010 |
| E |
0.112 |
0.103 |
0.094 |
| Remainder |
0.59 |
0.64 |
0.66 |
DFT calculations were performed to predict the geometry of the neutral, radical anion and dianion states of the DI[n]Ts. For computational ease, trimethylsilyl was used in place of the larger TIPS/TES groups. Bond distances in the core of DI2T are shown in Fig. 10 and Table S3;‡ see ESI for calculated geometries of DI1T (Fig. S10 and Table S2‡) and DI3T (Fig. S12 and Table S4‡). The calculations replicate the quinoidal character of the neutral DI[n]Ts but overestimate the degree of bond alternation in DI2T and DI3T by as much as 0.02 Å compared to the solid state data (Table 1). Upon one electron reduction, the quinoidal bonds begin to homogenize as the unpaired electron is delocalized over the system. The dianion shows a reversal of the quinoidal pattern and thus the expected rearomatization of the central thiophenes.25 In accord with the CV experiments, the dianionic, fully aromatic species is stabilized with respect to the neutral state.
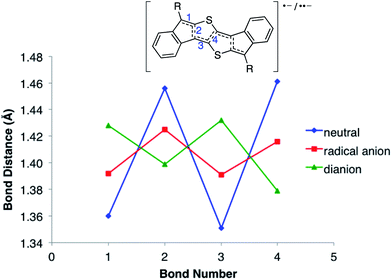 |
| | Fig. 10 DFT calculated DI2T bond distances upon reduction (R = (trimethylsilyl)ethynyl); performed at UCAM-B3LYP/6-31G(d,p) (neutral) and UCAM-B3LYP/6-31++G(d,p) (radical anion/dianion) level of theory. | |
Conclusions
In summary, the synthesis and characterization of a new class of quinoidal thienoacenes has been reported. The synthetic route is shown to be rapid and amenable to scale. X-ray crystallography corroborates the presence of distinct quinoidal motifs and reveals that the title compounds pack in progressively closer pairwise 1D arrangements. Analysis of the reduced states by EPR spectra and DFT calculations indicate stable anionic species. The large degree of π-orbital overlap, NIR absorption and favourable electrochemical properties suggest great potential for application in organic electronics. Future work will consist of further derivatization and expansion of the DI[n]T structures as well as testing their performance as organic semiconductors.
Acknowledgements
We thank the National Science Foundation (CHE-1301485) for support of this research as well as support in the form of instrumentation (CHE-0840478 and CHE-0923589) and computer grants (OCI-0960354). We also thank Dr Brad Rose for guidance on the EPR and computational studies and Prof. Mark Lonergan (University of Oregon) for use of his group's potentiostat. HRMS were obtained at the Mass Spectrometry Facilities and Services Core of the Environmental Health Sciences Center, Oregon State University, supported by grant #P30-ES00210, National Institute of Environmental Health Sciences, National Institutes of Health.
Notes and references
- J. Casado, R. Ponce Ortiz and J. T. López Navarrete, Chem. Soc. Rev., 2012, 41, 5672–5686 RSC.
- T. M. Pappenfus, R. J. Chesterfield, C. D. Frisbie, K. R. Mann, J. Casado, J. D. Raff and L. L. Miller, J. Am. Chem. Soc., 2002, 124, 4184–4185 CrossRef CAS PubMed.
- R. J. Chesterfield, C. R. Newman, T. M. Pappenfus, P. C. Ewbank, M. H. Haukaas, K. R. Mann, L. L. Miller and C. D. Frisbie, Adv. Mater., 2003, 15, 1278–1282 CrossRef CAS.
- T. Takahashi, K.-I. Matsuoka, K. Takimiya, T. Otsubo and Y. Aso, J. Am. Chem. Soc., 2005, 127, 8928–8929 CrossRef CAS PubMed.
- R. Ponce Ortiz, J. Casado, V. Hernández, J. T. López Navarrete, P. M. Viruela, E. Ortí, K. Takimiya and T. Otsubo, Angew. Chem., Int. Ed., 2007, 46, 9057–9061 CrossRef PubMed.
- S. Handa, E. Miyazaki, K. Takimiya and Y. Kunugi, J. Am. Chem. Soc., 2007, 129, 11684–11685 CrossRef CAS PubMed.
- Q. Wu, R. Li, W. Hong, H. Li, X. Gao and D. Zhu, Chem. Mater., 2011, 23, 3138–3140 CrossRef CAS.
-
(a) M. Lanata, C. Bertarelli, M. C. Gallazzi, A. Bianco, M. Del Zoppo and G. Zerbi, Synth. Met., 2003, 138, 357–362 CrossRef CAS;
(b) J. E. Raymond, J. Casado, J. T. López Navarrete, K. Takimiya and T. Goodson III, J. Phys. Chem. Lett., 2011, 2, 2179–2183 CrossRef CAS.
-
(a) I. Paci, J. C. Johnson, X. Chen, G. Rana, D. Popović, D. E. David, A. J. Nozik, M. A. Ratner and J. Michl, J. Am. Chem. Soc., 2006, 128, 16546–16553 CrossRef CAS PubMed;
(b) T. Minami and M. Nakano, J. Phys. Chem. Lett., 2012, 3, 145–150 CrossRef CAS.
-
A. G. Fix, D. T. Chase and M. M. Haley, in Topics in Current Chemistry, ed. J. S. Siegel and Y.-T. Wu, Springer, Berlin, Germany, DOI:10.1007/128_2012_376, in press.
-
(a) D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127–1130 CrossRef CAS PubMed;
(b) D. T. Chase, A. G. Fix, B. D. Rose, C. D. Weber, S. Nobusue, C. E. Stockwell, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 11103–11106 CrossRef CAS PubMed;
(c) D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed.
- A. G. Fix, P. E. Deal, C. L. Vonnegut, B. D. Rose, L. N. Zakharov and M. M. Haley, Org. Lett., 2013, 15, 1362–1365 CrossRef CAS PubMed.
- B. D. Rose, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Org. Lett., 2012, 14, 2426–2429 CrossRef CAS PubMed.
- B. S. Young, D. T. Chase, J. L. Marshall, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Chem. Sci., 2014, 5, 1008–1014 RSC.
-
(a) M. M. Payne, S. A. Odom, S. R. Parkin and J. E. Anthony, Org. Lett., 2004, 6, 3325–3328 CrossRef CAS PubMed;
(b) M. M. Payne, S. R. Parkin, J. E. Anthony, C.-C. Kuo and T. N. Jackson, J. Am. Chem. Soc., 2005, 127, 4986–4987 CrossRef CAS PubMed;
(c) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed;
(d) A. L. Briseno, Q. Miao, M.-M. Ling, C. Reese, H. Meng, Z. Bao and F. Wudl, J. Am. Chem. Soc., 2006, 128, 15576–15577 CrossRef CAS PubMed.
-
(a) J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed;
(b) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
-
(a) T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed;
(b) N. Nicolaus, P. T. Franke and M. Lautens, Org. Lett., 2011, 13, 4236–4239 CrossRef CAS PubMed.
- L. S. Fuller, B. Iddon and K. A. Smith, J. Chem. Soc., Perkin Trans. 1, 1997, 3465–3470 RSC.
- J. T. Henssler and A. J. Matzger, Org. Lett., 2009, 11, 3144–3147 CrossRef CAS PubMed.
- Crystallographic data for DI1T-TIPSE: C40H50SSi2, M = 619.04, 0.25 × 0.12 × 0.02 mm, T = 100 K, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.3989(9) Å, b = 12.4017(10) Å, c = 17.7936(19) Å, α = 88.626(6)°, β = 88.890(8)°, γ = 81.912(6)°, V = 1834.2(3) Å3, Z = 2, Dc = 1.121 Mg m−3, μ = 1.585 mm−1, F(000) = 668, 2θmax = 135.38°, 22289 reflections, 6439 independent reflections [Rint = 0.0405], R1 = 0.0395, wR2 = 0.0997 and GOF = 1.031 for 6439 reflections (388 parameters) with I > 2σ(I), R1 = 0.0511, wR2 = 0.1071 and GOF = 1.031 for all reflections, max/min residual electron density +0.383/−0.282 eÅ3. CCDC 1000352. Crystallographic data for DI1T-TESE: C34H38SSi2, M = 534.88, 0.29 × 0.06 × 0.02 mm, T = 100 K, triclinic, space group P, a = 7.0866(8) Å, b = 16.2124(19) Å, c = 26.849(3) Å, α = 85.812(10)°, β = 84.502(7)°, γ = 77.978(7)°, V = 2998.7(6) Å3, Z = 4, Z′ = 2, Dc = 1.185 Mg m−3, μ = 1.866 mm−1, F(000) = 1144, 2θmax = 120.0°, 24784 reflections, 8380 independent reflections [Rint = 0.0710], R1 = 0.1233, wR2 = 0.2892 and GOF = 1.130 for 8380 reflections (667 parameters) with I > 2σ(I), R1 = 0.1495, wR2 = 0.3028 and GOF = 1.130 for all reflections, max/min residual electron density +1.101/−0.687 eÅ3. CCDC 1000354. Crystallographic data for DI2T: C42H50S2Si2, M = 675.12, 0.17 × 0.04 × 0.02 mm, T = 100 K, triclinic, space group P, a = 7.4177(10) Å, b = 14.5332(16) Å, c = 18.922(2) Å, α = 73.889(8)°, β = 84.135(10)°, γ = 78.229(10)°, V = 1916.2(4) Å3, Z = 2, Dc = 1.170 Mg/m3, μ = 2.055 mm−1, F(000) = 724, 2θmax = 114.98°, 12023 reflections, 4765 independent reflections [Rint = 0.0398], R1 = 0.0530, wR2 = 0.1247 and GOF = 1.021 for 4765 reflections (415 parameters) with I > 2σ(I), R1 = 0.0738, wR2 = 0.1356 and GOF = 1.021 for all reflections, max/min residual electron density +0.400/−0.282 eÅ3. CCDC 1000353. Crystallographic data for DI3T: C44H50S3Si2, M = 731.20, 0.25 × 0.12 × 0.02 mm, T = 150 K, triclinic, space group P, a = 7.5217(4) Å, b = 14.0563(8) Å, c = 20.3360(11) Å, α = 73.188(4)°, β = 81.718(4)°, γ = 82.685(4)°, V = 2028.36(19) Å3, Z = 2, Dc = 1.197 Mg m−3, μ = 2.449 mm−1, F(000) = 780, 2θmax = 133.82°, 24367 reflections, 6946 independent reflections [Rint = 0.0496], R1 = 0.0493, wR2 = 0.1232 and GOF = 1.014 for 6946 reflections (452 parameters) with I > 2σ(I), R1 = 0.0642, wR2 = 0.1330 and GOF = 1.026 for all reflections, max/min residual electron density +0.479/−0.559 eÅ3.
, a = 8.3989(9) Å, b = 12.4017(10) Å, c = 17.7936(19) Å, α = 88.626(6)°, β = 88.890(8)°, γ = 81.912(6)°, V = 1834.2(3) Å3, Z = 2, Dc = 1.121 Mg m−3, μ = 1.585 mm−1, F(000) = 668, 2θmax = 135.38°, 22289 reflections, 6439 independent reflections [Rint = 0.0405], R1 = 0.0395, wR2 = 0.0997 and GOF = 1.031 for 6439 reflections (388 parameters) with I > 2σ(I), R1 = 0.0511, wR2 = 0.1071 and GOF = 1.031 for all reflections, max/min residual electron density +0.383/−0.282 eÅ3. CCDC 1000352. Crystallographic data for DI1T-TESE: C34H38SSi2, M = 534.88, 0.29 × 0.06 × 0.02 mm, T = 100 K, triclinic, space group P, a = 7.0866(8) Å, b = 16.2124(19) Å, c = 26.849(3) Å, α = 85.812(10)°, β = 84.502(7)°, γ = 77.978(7)°, V = 2998.7(6) Å3, Z = 4, Z′ = 2, Dc = 1.185 Mg m−3, μ = 1.866 mm−1, F(000) = 1144, 2θmax = 120.0°, 24784 reflections, 8380 independent reflections [Rint = 0.0710], R1 = 0.1233, wR2 = 0.2892 and GOF = 1.130 for 8380 reflections (667 parameters) with I > 2σ(I), R1 = 0.1495, wR2 = 0.3028 and GOF = 1.130 for all reflections, max/min residual electron density +1.101/−0.687 eÅ3. CCDC 1000354. Crystallographic data for DI2T: C42H50S2Si2, M = 675.12, 0.17 × 0.04 × 0.02 mm, T = 100 K, triclinic, space group P, a = 7.4177(10) Å, b = 14.5332(16) Å, c = 18.922(2) Å, α = 73.889(8)°, β = 84.135(10)°, γ = 78.229(10)°, V = 1916.2(4) Å3, Z = 2, Dc = 1.170 Mg/m3, μ = 2.055 mm−1, F(000) = 724, 2θmax = 114.98°, 12023 reflections, 4765 independent reflections [Rint = 0.0398], R1 = 0.0530, wR2 = 0.1247 and GOF = 1.021 for 4765 reflections (415 parameters) with I > 2σ(I), R1 = 0.0738, wR2 = 0.1356 and GOF = 1.021 for all reflections, max/min residual electron density +0.400/−0.282 eÅ3. CCDC 1000353. Crystallographic data for DI3T: C44H50S3Si2, M = 731.20, 0.25 × 0.12 × 0.02 mm, T = 150 K, triclinic, space group P, a = 7.5217(4) Å, b = 14.0563(8) Å, c = 20.3360(11) Å, α = 73.188(4)°, β = 81.718(4)°, γ = 82.685(4)°, V = 2028.36(19) Å3, Z = 2, Dc = 1.197 Mg m−3, μ = 2.449 mm−1, F(000) = 780, 2θmax = 133.82°, 24367 reflections, 6946 independent reflections [Rint = 0.0496], R1 = 0.0493, wR2 = 0.1232 and GOF = 1.014 for 6946 reflections (452 parameters) with I > 2σ(I), R1 = 0.0642, wR2 = 0.1330 and GOF = 1.026 for all reflections, max/min residual electron density +0.479/−0.559 eÅ3.
- RMS deviation from planarity for the dimesityl derivative of 4 is 0.094 Å and 0.186 Å for the two crystallographically independent molecules; see ref. 12.
- Crystallographic data for 9: C18H8SO2, M = 288.30, 0.16 × 0.12 × 0.08 mm, T = 100 K, orthorhombic, space group Pca21, a = 15.913(6) Å, b = 11.398(4) Å, c = 6.921(2) Å, V = 1255.4(8) Å3, Z = 4, Dc = 1.525 Mg m−3, μ = 0.258 mm−1, F(000) = 592, 2θmax = 52.0°, 5496 reflections, 2112 independent reflections [Rint = 0.0608], R1 = 0.0529, wR2 = 0.1001 and GOF = 1.038 for 2112 reflections (191 parameters) with I > 2σ(I), R1 = 0.0960, wR2 = 0.1145 and GOF = 1.038 for all reflections, the Flack parameter is 0.00(15), max/min residual electron density +0.308/−0.434 eÅ3. CCDC 1003348. Crystallographic data for 13: C20H8O2S2, M = 344.38, 0.18 × 0.04 × 0.02 mm, T = 100 K, monoclinic, space group P21/c, a = 12.983(4) Å, b = 3.9148(12) Å, c = 14.969(4) Å, β = 111.93(2)°, V = 705.7(4) Å3, Z = 4, Dc = 1.621 Mg m−3, μ = 3.500 mm−1, F(000) = 352, 2θmax = 135.36°, 3991 reflections, 1188 independent reflections [Rint = 0.0848], R1 = 0.0848, wR2 = 0.2128 and GOF = 1.034 for 1188 reflections (109 parameters) with I > 2σ(I), R1 = 0.1084, wR2 = 0.2292 and GOF = 1.034 for all reflections, max/min residual electron density +0.536/−0.543 eÅ3.
- H. Reiss and A. Heller, J. Phys. Chem., 1985, 89, 4207–4213 CrossRef CAS.
- K. Yui, H. Ishida, Y. Aso, T. Otsubo, F. Ogura, A. Kawamoto and J. Tanaka, Bull. Chem. Soc. Jpn., 1989, 62, 1547–1555 CrossRef CAS.
- X-ray crystallography reveals rearomatization of the central ring upon dianion formation of a 6,12-diethynylindeno[1,2-b]fluorene derivative; see: B. D. Rose, N. J. Sumner, A. S. Filatov, S. J. Peters, L. N. Zakharov, M. A. Petrukhina and M. M. Haley, J. Am. Chem. Soc., 2014, 136 DOI:10.1021/ja503870z , in press.
Footnotes |
| † Presented in part at the 15th International Symposium on Novel Aromatic Compounds, Taipei, Taiwan, July 29, 2013. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, computational details, and copies of 1H and 13C NMR spectra. CCDC 1000352–1000355 and 1003348, 1003349. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc01432d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a