 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of a cyclic 6 + 6 hexamer of D-biotin and formaldehyde†
Micke
Lisbjerg
,
Bo M.
Jessen
,
Brian
Rasmussen
,
Bjarne E.
Nielsen
,
Anders Ø.
Madsen
and
Michael
Pittelkow
*
Department of chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark. E-mail: pittel@kiku.dk
First published on 22nd April 2014
Abstract
The discovery of receptors using templated synthesis enables the selection of strong receptors from complex mixtures. In this contribution we describe a study of the condensation of D-biotin and formaldehyde in acidic water. We have discovered that halide anions template the formation of a single isomer of a 6 + 6 macrocycle. The macrocycle (biotin[6]uril) is water-soluble, chiral and binds halide anions (iodide, bromide and chloride) with selectivity for iodide in water, and it can be isolated on a gram scale in a one-pot reaction in 63% yield.
Introduction
Templated synthesis is an attractive strategy for the preparation of complex receptors for supramolecular chemistry.1 There is a profound difference between the complexity of the known naturally occurring systems that employ templated synthesis and the artificial systems that have been developed.2,3 The difficulty in de novo design of high-affinity receptors in water has inspired the implementation of thermodynamic templating for the selection and synthesis of receptors. This idea has gained popularity in recent years in particular through the selection-based supramolecular methodology of dynamic combinatorial chemistry.4 By connecting building blocks via reversible chemical bonds and allowing these to self-assemble in the presence of the template strong receptors for the given template are preferentially synthesised.5The curcurbit[n]uril series of macrocycles has gained significant popularity in the past years due to their ability to form strong host–guest complexes with a wide range of cationic guest molecules in water (Fig. 1a).6 Hemicucurbit[n]uril macrocycles (Fig. 1b) have been explored and they tend to bind anions rather than cations.7 The syntheses of cucurbit[n]urils and hemicucurbit[n]urils are acid mediated condensation reactions between a cyclic N,N′-dialkylurea and formaldehyde.
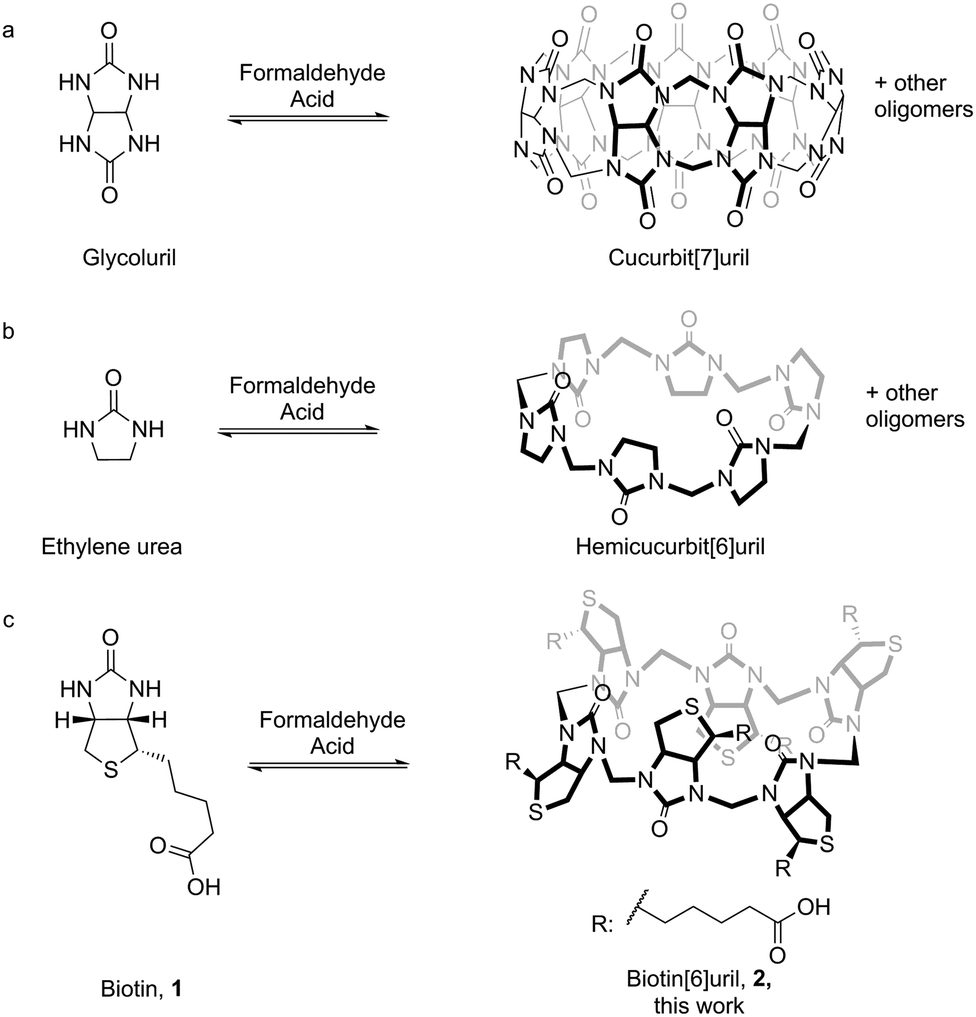 | ||
| Fig. 1 (a) Synthesis of cucurbit[n]uril by condensation of glycoluril and formaldehyde. (b) Synthesis of hemicucurbit[n]uril by condensation of ethylene urea and formaldehyde. (c) Synthesis of biotin[6]uril (2) by condensation of D-biotin and formaldehyde. | ||
Results and discussion
The resemblance between the structure of the starting ureas for the synthesis of cucurbit[n]urils and hemicucurbit[n]urils and the structure of D-biotin (1) inspired us to study the macrocyclisation reaction between D-biotin and formaldehyde (Fig. 1c). D-Biotin possesses a cyclic five-membered N,N′-dialkylurea that enables the oligomerisation reaction to form structures of the hemicucurbit[n]uril type. D-Biotin is known to form strong supramolecular complexes in water with the class of proteins called avidins,8 it has three chiral centers and the carboxylic acid functionality gives solubility in water.The acid-mediated reaction between D-biotin and formaldehyde in semi-concentrated hydrochloric acid at 60 °C resulted in the selective formation of a 6 + 6 macrocycle (biotin[6]uril, 2, Fig. 1c).9 In this macrocycle the six D-biotin units are connected through six methylene bridges. After optimisation of the reaction conditions in hydrochloric acid with respect to reaction times, temperature, and acid concentration, the product was isolated in 48% yield. We suspected that the choice of acid was important for the outcome of the reaction. Similar to aqueous HCl, the use of aqueous HBr and HI gave rise to the 6 + 6 macrocycle as the main product after 2 days of reaction at 60 °C (Fig. 2), whereas the use of sulfuric acid and otherwise similar reaction conditions resulted in the formation of a mixture of smaller non-cyclic oligomers. When chloride, bromide, or iodide salts were added to the sulfuric acid reaction mixture, the hexameric structure was again observed as the main product suggesting that a templating effect from the halide anions was the driving force for hexamer formation (Fig. 2). Using further optimised reaction conditions (3.5 M H2SO4 containing 5 M NaBr) 2 could be isolated in 63% yield on a multigram scale.
 | ||
| Fig. 2 HPLC chromatograms. Extracted ion chromatograms from LC/MS analyses of oligomers both cyclic and linear using different reaction mixtures. (I) 7 M HCl (the use of HBr and HI gave similar results), (II) 3.5 M H2SO4, (III) 3.5 M H2SO4 + 7 M NaCl, (IV) 3.5 M H2SO4 + 7 M NaBr and (V) isolated biotin[6]uril (2). The peak at 5 minutes corresponds to 2, all other peaks are smaller linear or cyclic products. | ||
The HPLC chromatogram of the isolated compound showed a single peak and the mass spectrum of the peak corresponded to a 6 + 6 macrocycle. Due to the non-equivalence of the two urea nitrogen atoms in D-biotin there are 9 different possible regioisomeric cyclic 6 + 6 hexamers (ESI, Fig. S11†). Analysis by 1H-NMR and 13C-NMR spectroscopy revealed the exclusive formation of a single species. In the NMR spectra of 2 (Fig. 3) only a single set of signals originating from D-biotin was observed. However, two signals resulting from two different aminal methylene bridges were present (between 4 and 4.5 ppm), as confirmed in the HSQC spectrum. The facts that there are two different methylene bridges, and that all six D-biotin units are identical enable us to unambiguously assign the structure as the C3-symmetrical isomer (Fig. 3).
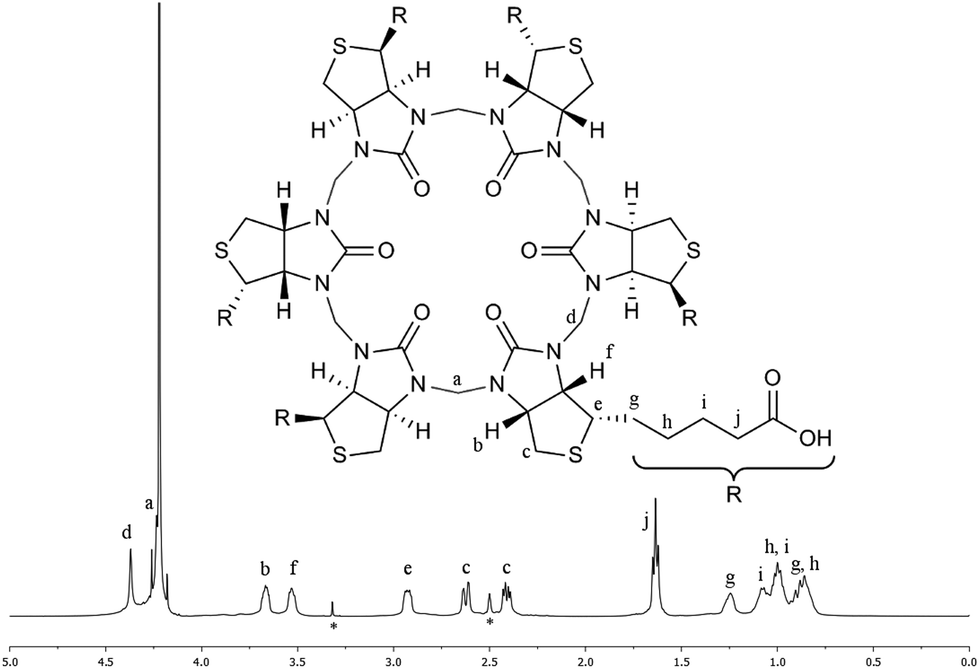 | ||
| Fig. 3 Characterisation of 2 by NMR spectroscopy. 1H-NMR spectrum of 2 in aqueous carbonate buffer with an external D6-DMSO standard (signals denoted with * originates from the D6-DMSO). The signal for (a) is situated on the edge of the water signal. All signals were unambiguously identified using a combination of 1H-, 13C-, COSY-, HMBC-, HCCOSW-, HSQC- and 1D-TOCSY NMR spectroscopy (ESI, Fig. S2–S9†). | ||
Elemental analysis of the product obtained when performing the macrocyclisation reaction in 7 M HCl (analysis for H, C, N and Cl) showed that 2 had precipitated as an HCl adduct. To investigate if the chloride anion was also encapsulated in the cavity of 2 in solution the complexation was investigated using NMR spectroscopy in aqueous carbonate buffer (as carbonate did not interact with 2). A series of 1H-NMR spectra were recorded at various concentrations of the 2-chloride complex. Upon dilution the C–H proton signals in the 1H-NMR spectrum of 2, which point towards the cavity in the crystal structure (see Fig. 4) were shifted significantly upfield. This exchange process between the bound and the unbound states were fast on the NMR chemical shift time scale. This suggests that the chloride anion was indeed interacting with the cavity of 2 in solution. When additional NaCl was added to the solution the same signals shifted in the opposite direction (downfield) (ESI, Fig. S10†).
 | ||
| Fig. 4 Single crystal X-ray structures of 2. (a and b) Side and top view of the crystal structure of 2 containing an ethanol molecule in the cavity. (c and d) Side and top view of the crystal structure of 2 encapsulating an iodide ion in the cavity. Yellow: sulphur; blue: nitrogen; gray: carbon; red: oxygen; purple: iodide; dark purple: sodium; hydrogens atoms and disordered atoms are omitted for clarity. | ||
The biotin[6]uril (2) was obtained chloride-free by treating the chloride inclusion complex with TlNO3 in alkaline solution followed by filtration and precipitation with sulfuric acid (the sulfate anion does not interact with the cavity of the macrocycle). The removal of the chloride anion was confirmed by dilution experiments, elemental analysis, and by obtaining the single crystal X-ray structure of the guest-free 2. The single crystal X-ray structure showed that the isolated product contained a solvent molecule (ethanol) in the cavity (Fig. 4a and b).
The structure of 2 has the D-biotin units situated in an alternating manner, with the uriedo moieties pointing in opposite directions with respect to the C3 symmetry axis of the macrocycle. The macrocycle has six side chains terminated with carboxylic acid groups, and in the crystal structure three side chains point in each direction as compared to the equator of the macrocycle. Each D-biotin moiety has a concave and a convex side, and in 2 all D-biotin units have the convex side with the two CH groups (protons b and f, Fig. 3) pointing into the centre of the macrocycle, giving an interior with twelve C–H groups.
We also succeeded growing crystals of an iodide inclusion complex suitable for single crystal X-ray analysis by co-crystallising 2 with NaI. Analysis of the crystal structure showed 2 with an iodide ion encapsulated in the cavity of the macrocycle and with the sodium ion sitting at the rim of the macrocycle coordinating to the carbonyl oxygen of one of the urea functionalities (Fig. 4c and d). The iodide–CH distances are slightly shorter than the sum of the van der Waals distances (shortest H–I distance in the crystal structure is 3.032 Å). This indicates a favourable interaction. The circumference of the cavity (measured via the six urea and the six methylene moieties) changes only from 33.541 Å with EtOH in the cavity to 33.693 Å with iodide in the cavity. The iodide thus fits well inside the cavity without altering the conformation significantly.
The binding properties of 2 towards the anions chloride, bromide and iodide were investigated in carbonate buffer (Fig. 5). Analysis of the binding stoichiometries using the continuous variation method (Job's method) for all the three halides indicated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometries (see ESI†). The binding affinities were determined by titration experiments using 1H-NMR spectroscopy and Isothermal Titration Calorimetry (ITC). When analysing the data from the 1H-NMR titrations it was convenient to monitor the chemical shift change of the C–H protons of the D-biotin units (protons b and f in Fig. 3), as these protons point towards the centre of the macrocyclic cavity, and experience a significant downfield shift upon halide binding.
:1 binding stoichiometries (see ESI†). The binding affinities were determined by titration experiments using 1H-NMR spectroscopy and Isothermal Titration Calorimetry (ITC). When analysing the data from the 1H-NMR titrations it was convenient to monitor the chemical shift change of the C–H protons of the D-biotin units (protons b and f in Fig. 3), as these protons point towards the centre of the macrocyclic cavity, and experience a significant downfield shift upon halide binding.
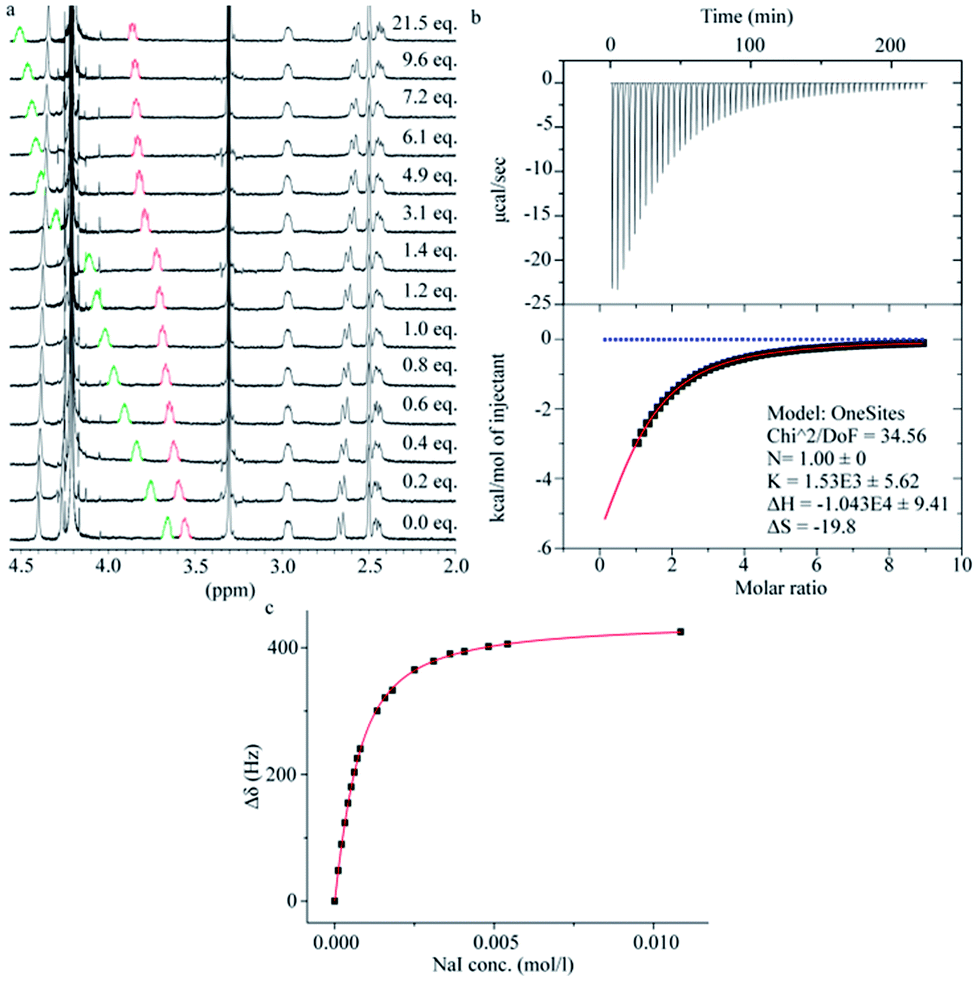 | ||
| Fig. 5 Binding studies between 2 and NaI. (a) Change in the 1H-NMR spectra of 2 upon addition of NaI. (b) Fitted and experimental data for the ITC titration between biotin[6]uril and iodide. Red: fitting; Blue: dilution. (c) Fitted and experimental data for the 1H-NMR titration between 2 and iodide. Measurements performed in water at pH 7.5. | ||
The binding strengths were in the order iodide > bromide > chloride, and we observed no binding to fluoride. The ITC data (Fig. 5b) showed the same order of binding constants as observed by the NMR titrations (Table 1) and furthermore gave a full picture of the thermodynamics of the binding events. The binding of the three halides have an unfavorable entropic (ΔS) contribution, but a favorable enthalpic (ΔH) contribution.
| Chlorideb | Bromideb | Iodideb | |
|---|---|---|---|
|
a Values for ΔH and ΔS were measured by ITC.
b The 1:1 stoichiometries of all complexes were confirmed by the continuous variation method (Job's plot). The uncertainty on the data is less than 10%.
|
|||
| K NMR (20 °C) | 59 M−1 | 5.4 × 102 M−1 | 2.2 × 103 M−1 |
| K ITC (30 °C) | 10 M−1 | 3.8 × 102 M−1 | 1.5 × 103 M−1 |
| ΔHa | −51.6 kJ mol−1 | −36.9 kJ mol−1 | −43.8 kJ mol−1 |
| ΔSa | −150.8 J mol−1 K−1 | −72.2 J mol−1 K−1 | −83.2 J mol−1 K−1 |
In order to examine the mode of formation, and thereby rationalise the high regioselectivity of the macrocyclisation reaction, we followed the progress of the reaction by LC/MS analysis. We performed a series of identical experiments in 7 M HCl at 60 °C (82 mM D-biotin, 5 equiv. formaldehyde). The reactions were stopped after different reaction times by quenching with base. In the early stages of the reaction D-biotin (1) and a linear dimer consisting of two D-biotins connected by one formaldehyde-based methylene bridge (3, Fig. 6) were the most abundant species. After 2–4 hours 2 began to form, and after 2–4 days it was the main product.
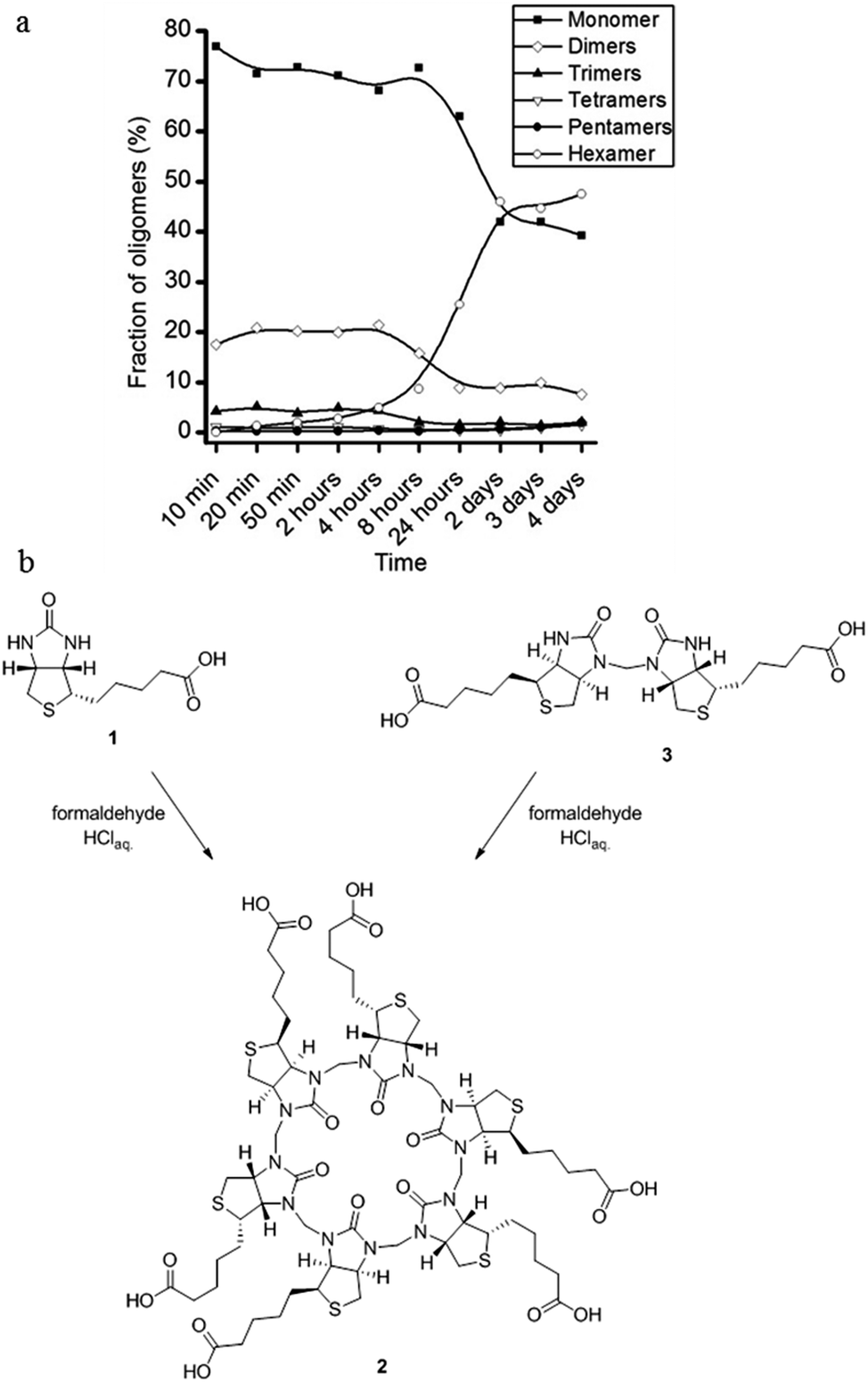 | ||
| Fig. 6 Following the reaction using LC/MS. (a) Evolution of the reaction between D-biotin and formaldehyde showing that only minor amounts of linear oligomers are detectable during the reaction. (b) Synthesis of 2 from D-biotin (1) and formaldehyde or from the D-biotin dimer (3) and formaldehyde give similar yields. | ||
This led us to the hypothesis that the hexamer was formed by a trimerisation of the linear dimer 3 with three additional equivalents of formaldehyde (Fig. 6). We found that the linear dimer (3) could be isolated by condensing two equivalents of D-biotin and one equivalent of formaldehyde in dilute H2SO4. We only observe the linear dimer (3) where the nitrogen positioned on the unsubstituted side of the D-biotin had reacted with the formaldehyde. When this dimer was subjected to the reaction conditions developed for the macrocyclisation reaction (7 M HCl, 5 equiv. formaldehyde), we also observe the formation of 2 in a similar yield.
Conclusions
To conclude we have discovered a 6 + 6 macrocyclic hexamer by condensation of D-biotin and formaldehyde that is chiral, water soluble and binds halide anions in water. The addition of halide anions (bromide) to the reaction mixture in H2SO4 gave 63% yield of the macrocycle. The selection of 2 is templated by halide anions and precipitation of the macrocycle–anion complex adds to the ease of isolation of biotin[6]uril.Acknowledgements
We acknowledge financial support from the Danish Research council (FNU) for a Steno Fellowship (MP), the Lundbeck Foundation for a Young Group Leader Fellowship (MP) and the Villum foundation for a diffractometer for determining the single crystal X-ray structures. The support and sponsorship provided by COST Action CM1005 is acknowledged. We thank Dr. Sophie R. Beeren for insightful discussions.Notes and references
- (a) S. Anderson, H. L. Anderson and J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469–475 CrossRef CAS; (b) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed.
- M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef CAS PubMed.
- F. Diederich and P. Stang, in Templated Organic Synthesis, Wiley-VCH, 2000 Search PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- S. Otto, Acc. Chem. Res., 2012, 45, 2200–2210 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- (a) Y. Miyahara, K. Goto, M. Oka and T. Inazu, Angew. Chem., Int. Ed., 2004, 43, 5019–5022 CrossRef CAS PubMed; (b) J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49, 2378–2381 CrossRef CAS PubMed; (c) R. Aav, E. Shmatova, I. Reile, M. Borissova, P. Topic and K. Rissanen, Org. Lett., 2013, 15, 3786–3789 CrossRef CAS PubMed.
- M. Wilchek, E. A. Bayer and O. Livnah, Immunol. Lett., 2006, 103, 27–32 CrossRef CAS PubMed.
- We call this family of macrocycles biotin[n]urils due to the resemblance to the cucurbit[n]uril class of macrocycles.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data and characterisation data: synthetic protocols, NMR titration data, single crystal X-ray crystallography data. CCDC 994261 and 988132. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc00990h |
| This journal is © The Royal Society of Chemistry 2014 |