 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precision polymers: a kinetic approach for functional poly(norbornenes)†
Dafni
Moatsou
,
Claire F.
Hansell
and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: R.K.O-Reilly@warwick.ac.uk
First published on 26th March 2014
Abstract
The potential control over monomer sequence in the ring-opening metathesis polymerization of functional norbornenes is explored based on the difference in reactivity of endo and exo isomers. This kinetic approach allows the rapid consumption of the exo-norbornene and insertion onto the growing poly(norbornene) chain within a narrow region of the overall polymer chain, whilst maintaining a homogeneous backbone. We herein demonstrate that this can be achieved using a range of functional monomers easily derived from commercially available precursors while their polymerization is carried out in a controlled manner.
Introduction
Efforts to synthesize sequence-controlled polymers are dedicated towards replicating the complex, yet precise, structures of macromolecules found in nature. It is anticipated that achieving even a fraction of nature's precision will lead to materials with finely tunable properties and functions.1,2 As much as these are extremely wide-ranging, they are all derived from subtle variations in the sequence of only a few building blocks. Whilst synthetic polymers are not a recent development, the progress in defined structures, compositions and sequences has been immense over the past few decades with the introduction of controlled polymerizations that allow the synthesis of complex copolymers such as block, graft, alternating and gradient materials.3Techniques that allow the sequential polymerization of functional monomers in a precise manner are still, however, somewhat limited.4 Exact control over the macromolecular sequence in polymeric materials tends to involve step-growth processes, whereby sequences are the result of the addition of one – and only one – monomer unit on the growing chain end by an iterative addition–activation process. Such processes include esterification,5 nitrone-mediated radical coupling,6 amidation,7,8 Horner–Wadsworth–Emmons (HWE) chemistry,9 Wittig olefination,10 the Passerini reaction,11 thiolactone chemistry12 and azide–alkyne Huisgen cycloaddition.13 A one-pot method was recently reported where the polymer consisted of a repeated short sequence that was first synthesized via a combination of organic reactions.14 While these step-growth approaches are highly reliable in terms of absolute sequence control, they are limited to the production of oligomers due to the complexity of the synthetic procedures and the cumulative effects of imperfect coupling, while the disproportionate high cost and poor product yield adds to the disadvantages. Perhaps the most efficient and widely used such technique is solid phase peptide synthesis,7,15 whereas other elegant approaches to sequence control, such as DNA templating,10,16 significantly suffer from scale and cost issues. This is a considerable drawback when contemplating the use of such methods for the exploration of materials with properties derived from their sequence.
A few attempts to harness control of the sequence in addition polymerizations (typically radical processes) can be found in the literature,17 but perhaps the most extensive study on “precision polymers”2 – that is macromolecules whose structure is more sharply defined than typical (co)polymers – has been conducted over the past few years by Lutz and co-workers.18 By taking advantage of the high reactivity of N-substituted maleimides towards styrenic monomers during their radical polymerization,19 a series of functionalities have been incorporated into a polystyrene backbone in a sequential manner. This strategy has been employed for the synthesis of polymers with a variety of pendent functionalities20 as well as to achieve more complex structures (i.e. graft, branch, dendritic polymers).21 Indeed, more recently Chan-Seng et al. have shown the possibility of further controlling the sequence of the polymer using automated synthetic protocols.22 These kinetic approaches are bridging the gap between selective and precise introduction of reactive functionalities and scalable and readily accessible polymeric materials. However, one fundamental drawback of the use of the styrenic–maleimide pair is the statistical, if not alternating,23 rather than precise monomeric, incorporation of the functional (maleimide) monomer at low conversion regimes of the auxiliary (styrenic) monomer; a solution to which has been shown to be time-controlled consecutive feeds of the two monomers, but one which adds a further layer of complexity to the synthetic approach.24
Ring-opening metathesis polymerization (ROMP) is a controlled polymerization method,25 the simplicity and versatility of which has allowed its use in industrial processes.26 In terms of available monomers, ROMP has been most successful with strained cyclic olefins, a major group of which are norbornenes (including oxa- and aza-norbornenes).27 While norbornene derivatives are easily synthesized bearing virtually unlimited possible functionalities, ROMP in the presence of certain functionalities is challenging due to catalyst poisoning and subsequent termination of the polymerization.28 Undeniably, the ability to synthesize nearly monodisperse polymers and complex architectures relies on the range of powerful catalysts available. These have allowed the synthesis of almost perfectly alternating copolymers based on the thermodynamically driven selective bond formation between the explored monomers.29 While these examples demonstrate the potential of ROMP in its use towards the synthesis of sequence-defined polymers, they are limited by the choice of monomer pairs and they often suffer from broad molecular weight distributions.
It has been shown that while exo-norbornenes rapidly undergo ROMP in the presence of ruthenium-based catalysts, endo-norbornenes exhibit far slower polymerization kinetics attributed primarily to steric interactions between the growing polymer chain and the incoming monomer.30 This has hereinto been perceived as a drawback to using an endo/exo monomer mixture as the overall polymerization rate decreases relative to a pure exo monomer feed,31 however we propose taking advantage of the slow polymerization of endo-norbornenes by using them as the auxiliary monomer in precision polymer synthesis. The introduction of a pure exo-norbornene monomer into the polymerization reaction should therefore result in its rapid insertion onto the growing polymer chain without otherwise altering the overall endo reaction rate. Given that the addition of exo monomers does not affect the “living” nature of the polymerization, this should achieve a short sequence of incorporated exo monomers at a specific position along the chain. In terms of exploring the material properties of precision polymers, a significant advantage of this system is the homogeneous copolymer backbone, due to the fact that the generic endo and exo monomers are stereoisomers.
In order to assess the relative polymerization rates of endo- and exo-norbornenes, N-hexyl-endo-norbornene-5,6-dicarboximide (endo-HexNb) was used as the auxiliary monomer and was copolymerized with three batches of 7-coumarinyl-exo-5-norbornene-2-carboxylate (exo-CoumNb) utilizing Grubbs’ 1st generation catalyst in chloroform at room temperature.‡ At different conversions of endo-HexNb, exo-CoumNb was introduced into the reaction mixture and the progress of the polymerization was monitored by 1H NMR spectroscopy (Fig. 1). It is apparent that despite the overall reaction being an exo–endo copolymerization, which might be expected to negatively affect the polymerization rate,31exo-CoumNb is consumed significantly faster than endo-HexNb. From the slopes of the regression lines of the ln([M]0/[M]) plots, the apparent polymerization rates (kapp) at different endo monomer conversions were determined. The overall polymerization rate of the endo monomer was found to be 2.68 × 10−6 s−1, and the obtained values for the exo monomer were found to be 2.26 × 10−4 s−1 at low endo monomer conversion (25%), 1.72 × 10−4 s−1 at 50% endo monomer conversion and 3.88 × 10−4 s−1 at higher conversion (70%), ∼100 times faster. This demonstrates that the rate of polymerization for the exo-CoumNb is independent of the conversion of endo-HexNb at which it was copolymerized, suggesting that precise insertion is possible throughout the polymerization.
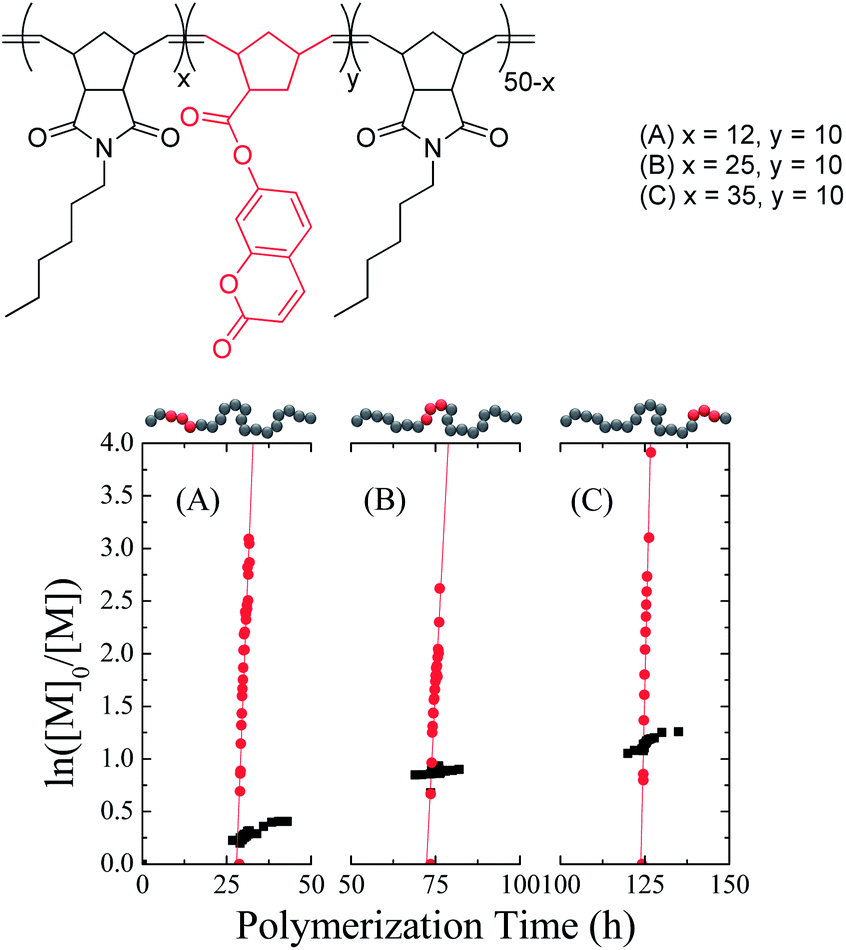 | ||
| Fig. 1 Chemical structure of the three copolymers (top) and relation between ln([M]0/[M]) and polymerization time (bottom) for the copolymerization of endo-HexNb (squares) and exo-CoumNb (circles) in CDCl3 at room temperature at three homopolymerization regimes: (A) 25%, (B) 50% and (C) 70% conversion of the endo monomer. | ||
A key issue that needs to be addressed when contemplating sequence control is the ability to insert the selected functionality on all growing polymer chains, while controlling the average equivalents added on each chain. To investigate how successful this was in the ROMP case, endo-HexNb was again used as the auxiliary monomer and exo-CoumNb as the functional monomer, one equivalent of which was inserted into the same polymer backbone at four different positions. The use of a coumarin-functionalized norbornene as the functional monomer provides a useful UV handle for characterization of the resultant copolymers. A sample was removed from the polymerization mixture before and after each addition of exo-CoumNb and was characterized by 1H NMR spectroscopy and SEC (size exclusion chromatography) analysis. Fig. 2 shows the progression of the molecular weight of the resultant copolymers with reaction time. The overall molecular weight increases and the molecular weight distribution decreases over a period of 355 h in a manner consistent with controlled/“living” polymerizations, suggesting that the polymerization kinetics and control of the auxiliary endo-norbornene are unperturbed by each single addition of the exo-norbornene.
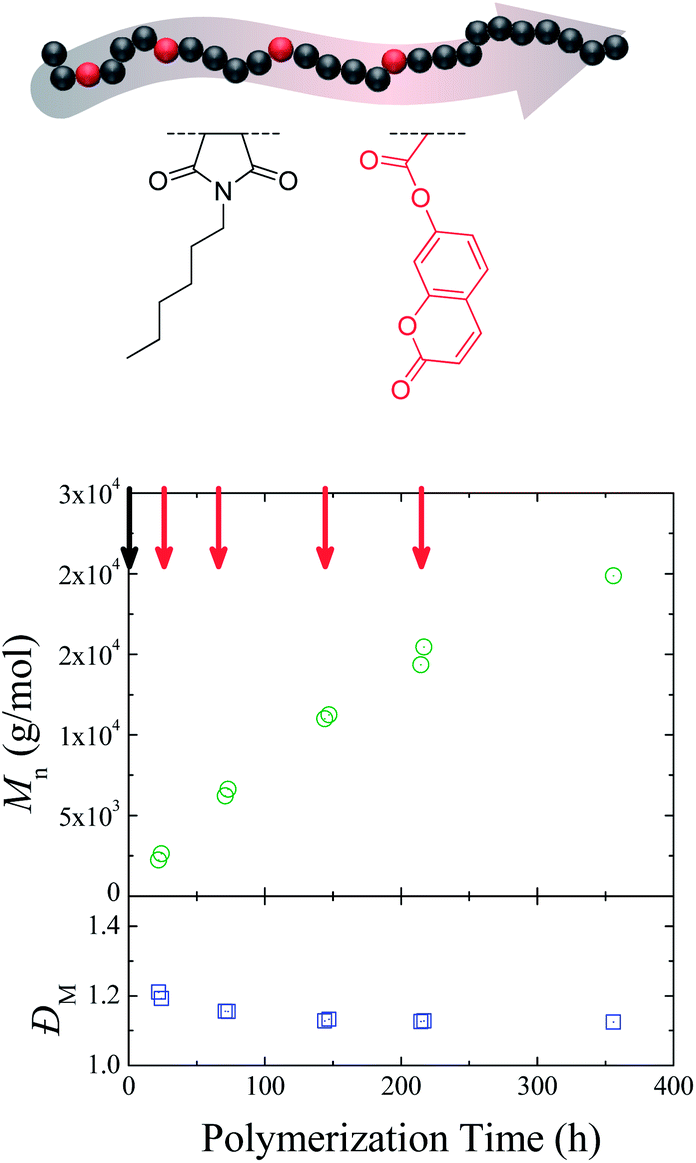 | ||
| Fig. 2 Chemical structure (top) and evolution of number-average molecular weight (Mn) and molecular weight distribution (ĐM) as a function of polymerization time (bottom) for the poly(endo-HexNb-co-exo-CoumNb). Red arrows denote single exo monomer addition. | ||
Fig. 3A shows the SEC traces of the final polymer (Mn = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1, ĐM = 1.12) using both a UV detector measuring the coumarin absorbance at 309 nm and a differential refractive index detector. Both traces exhibit identical distributions, which is indicative of successful incorporation of coumarin moieties in all growing polymer chains.
900 g mol−1, ĐM = 1.12) using both a UV detector measuring the coumarin absorbance at 309 nm and a differential refractive index detector. Both traces exhibit identical distributions, which is indicative of successful incorporation of coumarin moieties in all growing polymer chains.
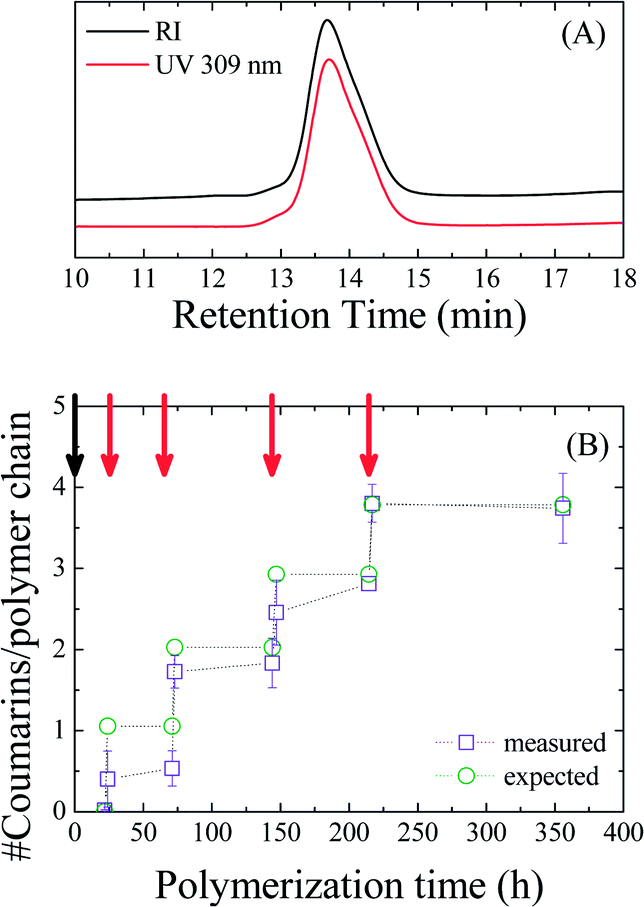 | ||
| Fig. 3 SEC traces of the final sample using a differential refractive index detector and a UV detector at 309 nm (A) and calculated amount of coumarin moieties per polymer chain vs. polymerization time (B) for the poly(endo-HexNb-co-exo-CoumNb) with addition points of a single exo monomer indicated by the red arrows. | ||
That being established, we attempted to quantify the average amount of coumarin moieties incorporated after each addition of exo-CoumNb by measuring UV-vis absorption spectra of the copolymer samples in CH2Cl2 (Fig. S3†). The concentration of coumarin in each sample was calculated based on the maximum coumarin absorption (between 310 and 320 nm) and was correlated to the overall polymer concentration. Fig. 3B shows the calculated average number of coumarin moieties per polymer chain with respect to polymerization time as calculated from the UV-vis measurements and is compared to the anticipated amount from the ratio of exo-CoumNb to the catalyst present in the reaction mixture. Ideally, after each monomer addition the ratio increases by one, thus suggesting the incorporation of one coumarin moiety per polymer chain on average. While at the initial stages of the polymerization the average coumarin content per polymer chain is low, suggesting incomplete incorporation of the exo monomer, upon the last addition, the measured average reaches the expected number of coumarin moieties per polymer chain. While this confirms that the average amount of exo monomers inserted per growing polymer chain is controlled, it also shows that the precision with which that monomer is inserted is limited. Further investigations into the factors that dictate the precision of the insertion will focus on the exo monomer steric shielding and the mechanistic aspects of cross-polymerization.
The versatility of ROMP in terms of synthesizing precision polymers was assessed by the sequential addition of four different functional exo-norbornenes (2 eq. each) in the polymerization mixture of endo-HexNb at different monomer conversions. The simplicity of the monomer synthesis afforded the selection of four exo-norbornenes bearing diverse functionalities to provide the polymer with unique properties: pentafluorophenyl exo-5-norbornene-2-carboxylate (exo-PFPNb), (1-pyrenyl)methyl exo-5-norbornene-2-carboxylate (exo-PyrNb), (trimethylsilanyl)methyl exo-5-norbornene-2-carboxylate (exo-TMSNb) and the previously mentioned exo-CoumNb. The incorporation of each functionality into the growing polymer chain was monitored by 1H NMR spectroscopy while the polymerization rates were calculated from the slopes of the regression lines of the ln([M]0/[M]) plots (Fig. 4A). Fig. 4B shows the SEC traces of the final polymer (Mn = 7750 g mol−1, ĐM = 1.16) using a UV detector measuring the coumarin absorbance at 309 nm and the pyrene absorbance at 344 nm as well as a differential refractive index detector. All traces exhibit identical narrow distributions, suggesting good control of the polymerization as well as the presence of both coumarin and pyrene moieties in all growing polymer chains.
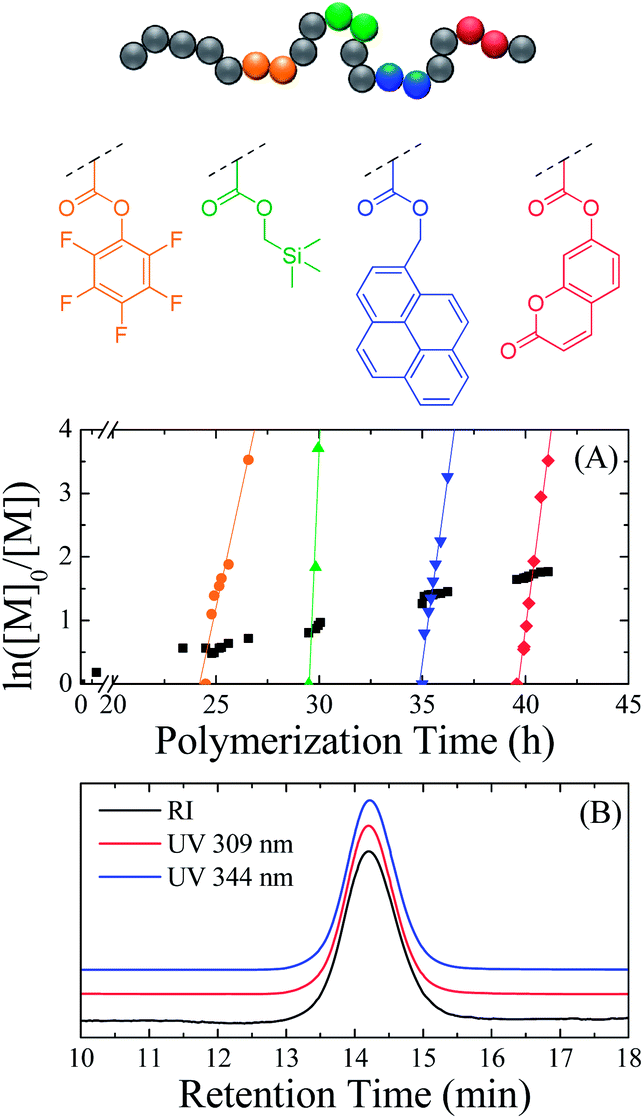 | ||
| Fig. 4 Relation between ln([M]0/[M]) and polymerization time (A) and SEC traces of the final copolymer using a differential refractive index detector and a UV detector at 309 nm and 344 nm (B) for the copolymerization of endo-HexNb and exo-norbornenes with different functionalities. | ||
Once again, the overall polymerization rate of the auxiliary endo monomer was low while each exo-norbornene was copolymerized significantly faster. Interestingly, the kapp of endo-HexNb was found to be 1.43 × 10−5 s−1 which is higher than in our previous experiments, and the kapp of the exo-norbornenes were found to be 4.16 × 10−4 s−1 for exo-PFPNb, 2.12 × 10−3 s−1 for exo-TMSNb, 6.56 × 10−4 s−1 for exo-PyrNb and 6.94 × 10−4 s−1 for exo-CoumNb which is lower than previously seen. This is possibly a result of the different relative concentrations of the reagents – i.e. while the endo-norbornene concentration is comparable, the catalyst concentration is higher whereas the exo-norbornene concentration is lower. It should also be noted that exo-TMSNb exhibited a significantly higher polymerization rate compared to the other exo monomers which was attributed to the less bulky pendent group and suggests that relative reaction rates could be easily tuned by adjusting the steric bulk of the endo and exo monomers. Nevertheless, the polymerization of the exo monomers was over 30 times faster than that of the auxiliary monomer indicating the successful synthesis of a multifunctional precision polymer through sequential monomer addition.
Conclusions
To conclude, we have demonstrated the synthesis of polymers via ROMP with the ability to easily and relatively accurately control the position of functional moieties on the polymer chain, by taking advantage of the vastly different kinetics of endo- and exo-norbornenes. It was shown that when adding exo monomer in small, defined batches to the auxiliary endo polymerization, the presence of both monomers had little effect on their respective reactivity as the endo-norbornene was consumed 30–100 times slower than the exo-norbornene during their copolymerization. The accuracy of the monomer addition was investigated by sequentially inserting an exo-norbornene bearing a UV-active functionality into a growing endo chain at the desired polymerization conversions. In addition, we have successfully synthesized a polymer containing four different functionalities at relatively known positions on the backbone. We believe that any isomerically pure norbornenes that can undergo ROMP can be used as building blocks for the synthesis of precision polymers following this method, expanding the library of possible precision polymers. As ROMP is simple to carry out, can be performed with a range of monomer functionalities, results in a homogeneous backbone and is entirely kinetically driven, we believe that this work complements the precision polymer strategy followed by the Lutz group in leading to a new class of materials with programmable compositions.Acknowledgements
The EPSRC and University of Warwick are acknowledged for funding. Some of the equipment used in this research was obtained through Birmingham Science City with support from Advantage West Midlands and partially funded by the European Regional Development Fund. The authors would like to thank Mr Edward Tunnah for his assistance with NMR spectroscopy, and Dr Helen Willcock for proof-reading.Notes and references
- (a) N. Badi and J.-F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC; (b) J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC; (c) R. Jones, Nat. Nanotechnol., 2008, 3, 699–700 CrossRef CAS PubMed.
- H. G. Börner, Macromol. Rapid Commun., 2011, 32, 115–126 CrossRef PubMed.
- E. Saldívar-Guerra and E. Vivaldo-Lima, in Handbook of Polymer Synthesis, Characterization, and Processing, John Wiley & Sons, Inc., 2013, pp. 1–14 Search PubMed.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- R. M. Stayshich, R. M. Weiss, J. Li and T. Y. Meyer, Macromol. Rapid Commun., 2011, 32, 220–225 CrossRef CAS PubMed.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2118–2126 CrossRef CAS.
- L. Hartmann and H. G. Börner, Adv. Mater., 2009, 21, 3425–3431 CrossRef CAS PubMed.
- (a) L. Hartmann, E. Krause, M. Antonietti and H. G. Börner, Biomacromolecules, 2006, 7, 1239–1244 CrossRef CAS PubMed; (b) L. Hartmann, Macromol. Chem. Phys., 2011, 212, 8–13 CrossRef CAS; (c) S. Mosca, F. Wojcik and L. Hartmann, Macromol. Rapid Commun., 2011, 32, 197–202 CrossRef CAS PubMed; (d) D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Biomacromolecules, 2012, 13, 1845–1852 CrossRef CAS PubMed; (e) F. Wojcik, S. Mosca and L. Hartmann, J. Org. Chem., 2012, 77, 4226–4234 CrossRef CAS PubMed.
- B. N. Norris, S. Zhang, C. M. Campbell, J. T. Auletta, P. Calvo-Marzal, G. R. Hutchison and T. Y. Meyer, Macromolecules, 2013, 46, 1384–1392 CrossRef CAS.
- P. J. Milnes, M. L. McKee, J. Bath, L. Song, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Chem. Commun., 2012, 48, 5614–5616 RSC.
- (a) S. C. Solleder and M. A. R. Meier, Angew. Chem., Int. Ed., 2014, 53, 711–714 CrossRef CAS PubMed; (b) A. Lv, X.-X. Deng, L. Li, Z.-L. Li, Y.-Z. Wang, F.-S. Du and Z.-C. Li, Polym. Chem., 2013, 4, 3659–3662 RSC.
- P. Espeel, L. L. G. Carrette, K. Bury, S. Capenberghs, J. C. Martins, F. E. Du Prez and A. Madder, Angew. Chem., Int. Ed., 2013, 52, 13261–13264 CrossRef CAS PubMed.
- (a) S. Pfeifer, Z. Zarafshani, N. Badi and J.-F. Lutz, J. Am. Chem. Soc., 2009, 131, 9195–9197 CrossRef CAS PubMed; (b) M.-A. Berthet, Z. Zarafshani, S. Pfeifer and J.-F. Lutz, Macromolecules, 2009, 43, 44–50 CrossRef.
- J.-J. Yan, D. Wang, D.-C. Wu and Y.-Z. You, Chem. Commun., 2013, 49, 6057–6059 RSC.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- (a) Y. He and D. R. Liu, Nat. Nanotechnol., 2010, 5, 778–782 CrossRef CAS PubMed; (b) M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Angew. Chem., Int. Ed., 2010, 49, 7948–7951 CrossRef CAS PubMed; (c) C. T. Calderone and D. R. Liu, Angew. Chem., Int. Ed., 2005, 44, 7383–7386 CrossRef CAS PubMed; (d) Z. J. Gartner, M. W. Kanan and D. R. Liu, J. Am. Chem. Soc., 2002, 124, 10304–10306 CrossRef CAS PubMed; (e) Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed; (f) Y.-Z. Ke, R.-J. Ji, T.-C. Wei, S.-L. Lee, S.-L. Huang, M.-J. Huang, C.-H. Chen and T.-Y. Luh, Macromolecules, 2013, 46, 6712–6722 CrossRef CAS.
- (a) A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed; (b) G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 Search PubMed; (c) K. Nakatani, Y. Ogura, Y. Koda, T. Terashima and M. Sawamoto, J. Am. Chem. Soc., 2012, 134, 4373–4383 CrossRef CAS PubMed; (d) Y. Hibi, S. Tokuoka, T. Terashima, M. Ouchi and M. Sawamoto, Polym. Chem., 2011, 2, 341–347 RSC; (e) S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808–10809 CrossRef CAS PubMed; (f) R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed; (g) Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS; (h) A. Natalello, J. N. Hall, E. A. L. Eccles, S. M. Kimani and L. R. Hutchings, Macromol. Rapid Commun., 2011, 32, 233–237 CrossRef CAS PubMed.
- J.-F. Lutz, Acc. Chem. Res., 2013, 46, 2696–2705 CrossRef CAS PubMed.
- S. Pfeifer and J.-F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS PubMed.
- (a) S. Srichan, D. Chan-Seng and J.-F. Lutz, ACS Macro Lett., 2012, 1, 589–592 CrossRef CAS; (b) J.-F. Lutz, B. V. K. J. Schmidt and S. Pfeifer, Macromol. Rapid Commun., 2011, 32, 127–135 CrossRef CAS PubMed; (c) N. Baradel, S. Fort, S. Halila, N. Badi and J.-F. Lutz, Angew. Chem., Int. Ed., 2013, 52, 2335–2339 CrossRef CAS PubMed; (d) R. Kakuchi, M. Zamfir, J.-F. Lutz and P. Theato, Macromol. Rapid Commun., 2012, 33, 54–60 CrossRef CAS PubMed; (e) S. Srichan, L. Oswald, M. Zamfir and J.-F. Lutz, Chem. Commun., 2012, 48, 1517–1519 RSC; (f) S. Pfeifer and J.-F. Lutz, Chem. Eur. J., 2008, 14, 10949–10957 CrossRef CAS PubMed.
- (a) B. V. K. J. Schmidt, N. Fechler, J. Falkenhagen and J.-F. Lutz, Nat. Chem., 2011, 3, 234–238 CrossRef CAS PubMed; (b) A. O. Moughton, T. Sagawa, W. M. Gramlich, M. Seo, T. P. Lodge and M. A. Hillmyer, Polym. Chem., 2013, 4, 166–173 RSC; (c) N. Baradel, O. Gok, M. Zamfir, A. Sanyal and J.-F. Lutz, Chem. Commun., 2013, 49, 7280–7282 RSC; (d) M. Zamfir, P. Theato and J.-F. Lutz, Polym. Chem., 2012, 3, 1796–1802 RSC.
- D. Chan-Seng, M. Zamfir and J.-F. Lutz, Angew. Chem., Int. Ed., 2012, 124, 12420–12423 CrossRef.
- (a) G.-Q. Chen, Z.-Q. Wu, J.-R. Wu, Z.-C. Li and F.-M. Li, Macromolecules, 1999, 33, 232–234 CrossRef; (b) K. Ishizu, C. Takashimizu, T. Shibuya and S. Uchida, Polym. Int., 2003, 52, 1010–1015 CrossRef CAS.
- M. Zamfir and J.-F. Lutz, Nat. Commun., 2012, 3, 1138 CrossRef.
- (a) C. Slugovc, Macromol. Rapid Commun., 2004, 25, 1283–1297 CrossRef CAS; (b) A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- (a) M. S. Trimmer, in Handbook of Metathesis, Wiley-VCH Verlag GmbH, 2008, pp. 407–418 Search PubMed; (b) M. Yamazaki, J. Mol. Catal. A: Chem., 2004, 213, 81–87 CrossRef CAS; (c) J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- M. R. Buchmeiser, Chem. Rev., 2000, 100, 1565–1604 CrossRef CAS PubMed.
- (a) A. Song, K. A. Parker and N. S. Sampson, J. Am. Chem. Soc., 2009, 131, 3444–3445 CrossRef CAS PubMed; (b) J. Romulus, L. Tan, M. Weck and N. S. Sampson, ACS Macro Lett., 2013, 2, 749–752 CrossRef CAS PubMed; (c) K. Vehlow, M. Lichtenheldt, D. Wang, S. Blechert and M. R. Buchmeiser, Macromol. Symp., 2010, 296, 44–48 CrossRef CAS; (d) M. Bornand and P. Chen, Angew. Chem., Int. Ed., 2005, 44, 7909–7911 CrossRef PubMed; (e) S. Torker, A. Müller and P. Chen, Angew. Chem., Int. Ed., 2010, 49, 3762–3766 CrossRef CAS PubMed; (f) M. Bornand, S. Torker and P. Chen, Organometallics, 2007, 26, 3585–3596 CrossRef CAS; (g) M. Abbas, J. Wappel and C. Slugovc, Macromol. Symp., 2012, 311, 122–125 CrossRef CAS; (h) T.-L. Choi, I. M. Rutenberg and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 3839–3841 CrossRef CAS.
- (a) V. Lapinte, J.-C. Brosse and L. Fontaine, Macromol. Chem. Phys., 2004, 205, 824–833 CrossRef CAS; (b) J. D. Rule and J. S. Moore, Macromolecules, 2002, 35, 7878–7882 CrossRef CAS; (c) M. Vrabel, P. Kole, K. M. Brunner, V. Lopez-Carrillo, R. de Vivie-Riedle and T. Carell, Angew. Chem., Int. Ed., 2013, 40, 13309–13312 Search PubMed.
- J. M. Pollino, L. P. Stubbs and M. Weck, Macromolecules, 2003, 36, 2230–2234 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional characterization data is included. See DOI: 10.1039/c4sc00752b |
| ‡ Initial attempts to use two isomers of the same monomer were unfruitful as we found that overlapping signals in the 1H NMR spectra significantly affected the accuracy with which we were able to analyze the monomer conversion and polymer composition. However, we have been able to confirm that exo-HexNb homopolymerization is as efficient as the exo-norbornenes shown in the manuscript (see ESI†). |
| This journal is © The Royal Society of Chemistry 2014 |