 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single-molecule spectroscopy of photosynthetic proteins in solution: exploration of structure–function relationships
Gabriela S.
Schlau-Cohen
a,
Samuel
Bockenhauer†
ab,
Quan
Wang
ac and
W. E.
Moerner
*a
aDepartment of Chemistry, Stanford University, Stanford, California, USA. E-mail: wmoerner@stanford.edu
bDepartment of Physics, Stanford University, Stanford, California, USA
cDepartment of Electrical Engineering, Stanford University, Stanford, California, USA
First published on 15th April 2014
Abstract
In photosynthetic light harvesting, absorbed photoenergy transfers through networks of pigment–protein complexes to a central location, known as the reaction center, for conversion to chemical energy. This process occurs with remarkable near-unity quantum efficiency. Pigment–protein complexes exhibit emergent properties very different from those of their component molecules, resulting from interactions among the pigments and between the pigments and the surrounding protein environment. Thus, the precise molecular mechanisms of the energy transport process are complex and coupled, obscuring their molecular origin. Furthermore, because these interactions are sensitive to the molecular structure, they vary from complex to complex and vary in time for each individual complex due to fluctuations of the protein's conformation. To explore the effect of complex-to-complex variation, we study individual photosynthetic proteins, one at a time. We use a solution-phase, single-molecule technique, known as the Anti-Brownian Electrokinetic (ABEL) trap, to elucidate the conformational dynamics of single photosynthetic pigment–protein complexes without introducing additional perturbations from immobilization strategies. After reviewing the principles of the ABEL trap, we demonstrate its application to the study of several photosynthetic pigment–protein complexes. We demonstrate that the ABEL trap approach can lead to an increased understanding of photosynthetic complexes by presenting three examples: (1) analysis of photodegradation pathways, (2) characterization of complex-to-complex heterogeneity, and (3) identification of distinct functional forms.
Introduction
Photosynthetic systems flourish by simultaneously balancing two tasks: (1) conversion of photoenergy to chemical energy with up to near-unity quantum efficiency; and (2) adaption to varying light intensities by safely dissipating excess photoenergy.1 These processes occur in networks of pigment–protein complexes that absorb sunlight and transfer the photoenergy from complex to complex to reach a central location, known as the reaction center, where charge separation occurs. Pigment–pigment and pigment–protein couplings within each complex drive these initial absorption and energy transport processes because they determine the excited state energies and dynamics. These couplings are highly sensitive to inter-molecular distances; they vary drastically with small changes in the protein structure. As a result, the excited state energies and dynamics vary from protein to protein due to differences in protein conformation and vary in one protein over time due to protein fluctuations.2,3 In ensemble measurements, the excited state energies and dynamics can be obscured because only the average values are observed. In single-molecule measurements, however, the static and dynamic heterogeneity in these properties can be explored.4–11Pigment–protein photosynthetic complexes typically consist of an array of pigments, specifically (bacterio)chlorophyll, carotenoids, and phycobilins, surrounded by a protein matrix. One well-studied pigment–protein complex is the primary antenna complex from purple bacteria, light-harvesting complex 2 (LH2). A structural model of LH2 is shown in Fig. 1a. It has a nonameric ring structure, where each monomer (shown in Fig. 1b) contains three bacteriochlorophyll (BChl) molecules and one carotenoid.12 The protein structure gives rise to two concentric rings of BChl, known as the B800 and B850 rings, based on their absorption maxima as shown in the linear absorption spectrum (Fig. 1c). The linear absorption spectrum consists of broad, well-separated peaks across the visible and near-infrared. Disentangling the molecular origin of the excited states giving rise to these peaks is challenging because the states that absorb and transfer photoenergy are shaped by the transitions of the uncoupled pigments, pigment–pigment interaction, and multiple types of interactions between pigments and the surrounding protein. Finally, inhomogeneity, or differences from protein to protein, further broadens these peaks in ensemble measurements. For example, whether the inhomogeneity arises from ellipticity in the protein ring or from subunit-to-subunit disorder has been debated.13,14 Overall, however, much has been learned from a variety of previous theoretical, biochemical, steady-state, and transient absorption studies.2,7,15,16
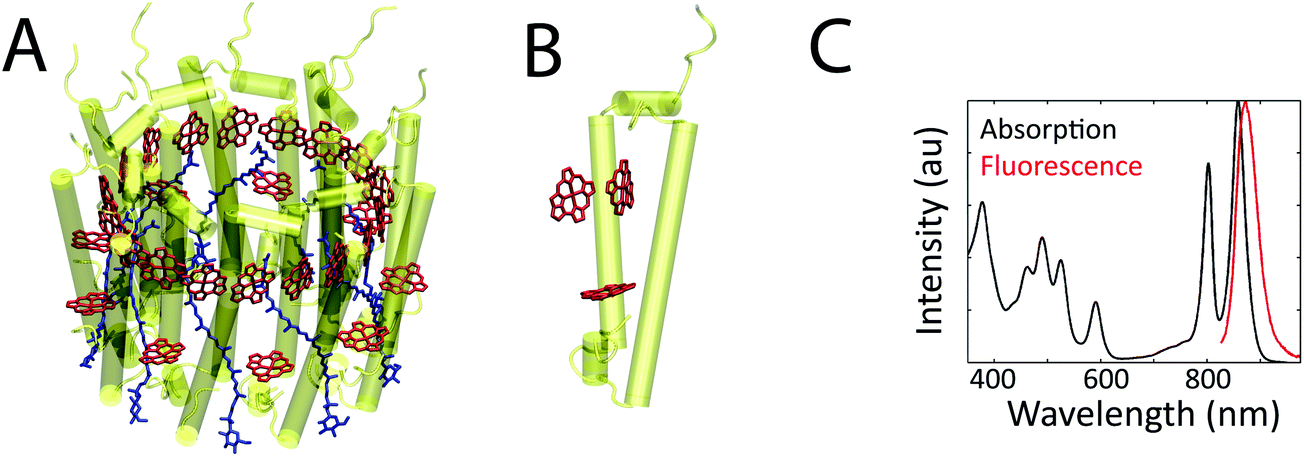 | ||
| Fig. 1 (a) The structural model from X-ray crystallography of the bacterial antenna pigment–protein complex, LH2, from R. acidophila (1KZU, Protein Data Bank). LH2 consists of nine subunits, arranged in a ring. (b) The protein and BChl for one subunit of LH2. The pigments are a few angstroms from each other and from the surrounding protein, which produces coupling between the pigments and between the pigments and the protein. (c) The ensemble linear absorption and fluorescence spectra of LH2 at room temperature. | ||
Single-molecule fluorescence spectroscopy has provided an incisive tool to characterize inhomogeneity and reveal information about the molecular origins of the excited states of pigment–protein complexes.7 Fluorescence can be a background-free measurement, and thus has the sensitivity to report on even single photosynthetic proteins. Both experimental and theoretical work has shown protein-to-protein variation in dynamics at the single-molecule level. In experimental work, single-molecule spectra of LH2 showed heterogeneity of emission energies,17–19 as well as symmetry-destroying fluctuations of the protein ring.13 Single-molecule spectra of the major light-harvesting complex of photosystem II (LHCII) from higher plants also exhibited heterogeneities in emission energy, which were attributed to conformational changes that introduced mixing with charge transfer states.20 Recent single-molecule ultrafast experiments observed heterogeneity in the excited-state dynamics, specifically in the periods and phases of quantum coherent oscillations in the first ∼400 fs after excitation.21 In theoretical work, heterogeneity in excited-state dynamics has also been observed, in both coherent oscillations22 and energy and electron transfer.23
The previous single-molecule work, however, all relied on various immobilization schemes that can perturb protein conformation.24–26 Because the pigment–protein interaction depends sensitively on the protein structure, this type of perturbation can alter the energies and dynamics of the excited states of pigment–protein complexes. In fact, conflicting fluorescence emission intensities and spectra have been attributed to differences in immobilization schemes.14
The Anti-Brownian Electrokinetic (ABEL) trap is a novel single-molecule tool that enables extended observation of single biomolecules in solution.4,27–31 By applying this technique to photosynthetic proteins, the perturbation from immobilization or encapsulation is removed, and the native inhomogeneity and dynamics can be explored. Application of the ABEL trap to photosynthetic systems has provided insight into the structure–function relationships that underlie the excited states of these systems.32–34 Here, we review experiments illustrating that the combination of a solution-phase environment and a single-molecule approach reveals hidden excited state characteristics and dynamics.
The principles of the ABEL trap
The ABEL trap holds single, solution-phase fluorescent particles in a micron-scale observation region for tens of seconds without perturbative attachment or encapsulation. Electrokinetic forces counter diffusion in the x–y plane.28 A microfluidic cell made of fused silica or PDMS and glass confines the particle to hundreds of nanometers in the z direction, which is less than the focal depth of an optical microscope.28The principle of the ABEL trap is a closed-loop feedback system, as illustrated in Fig. 2. Fluorescent particles are maintained within an observation region through three steps: (1) optical detection of position. The laser beam is constantly scanning in a micron-scale grid pattern at ∼kHz, which defines the observation region. When a fluorescent particle diffuses into the observation region and spatially and temporally overlaps with the beam, the particle can emit a photon. When a photon is detected, the position of the particle can be approximately inferred from the known position of the beam. (2) Electric fields are applied in x and y to the highly dilute (∼picomolar) sample solution held in a microfluidic cell. (3) The electric fields induce electrokinetic forces35 that move the particle back to the center of the observation region. This cycle is repeated for every detected photon to maintain the position of the fluorescent particle within the observation region until it enters a dark state, at which point no position information can be extracted and the particle diffuses away.28 Of particular note is the fact that the particle is trapped in a region of time-averaged uniform intensity. This means that on time scales longer than the beam motion, the molecule's brightness (intensity), a critical parameter for fluorescence analysis, can be precisely measured, as opposed to the case with fluorescence correlation spectroscopy (FCS36) where molecules diffuse randomly through a non-uniform Gaussian-shaped focal spot, requiring complex analysis and averaging to extract intensity information.37
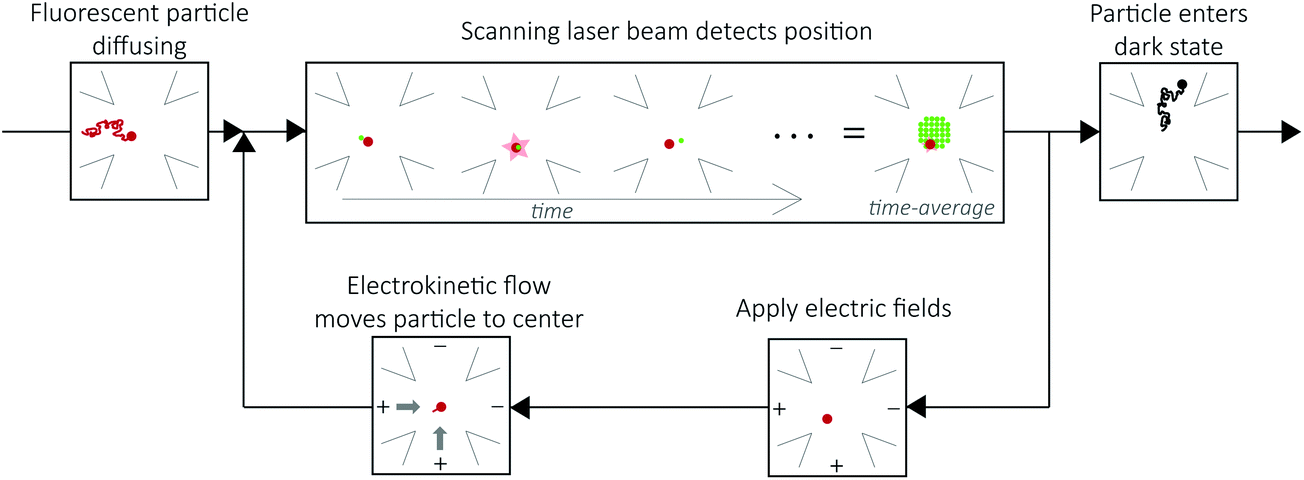 | ||
| Fig. 2 The ABEL trap functions through a closed loop feedback system. A fluorescent particle (red) diffuses into the observation region (upper left panel). The laser beam (green) is constantly scanning the observation region (upper center panel). When the laser beam overlaps with the particle, as shown, it can emit a photon. Upon detection of the photon, the position of the particle is approximated by the known position of the laser beam. Electric fields are applied (bottom right) along two orthogonal directions to induce electrokinetic flow to move the particle to the center of the observation region (bottom left). This cycle is repeated until the particle enters a dark state and diffuses away (upper right panel). | ||
Recent further technological improvements in the ABEL trap use a priori knowledge about particle dynamics to optimally estimate position (i.e. the Kalman filter) and apply feedback.29,31,38 This has enabled trapping of particles as small as single fluorophores, and thus demonstrates the ability to trap any soluble, fluorescently-labeled particle.30,31 This improved ability to trap molecules enables not just smaller particles, but also dimmer ones, to be studied. The ability to maintain dimmer particles within the field of view is particularly important for studying photosynthetic systems, as these experiments rely on the intrinsic fluorescence of the pigments inside the proteins, as opposed to artificially attached synthetic dyes specially designed to be stable single-molecule emitters.
In the ABEL trap, the emitted fluorescence intensity, lifetime, and spectrum can be simultaneously monitored for each single molecule.30 Changes in fluorescence intensity arise from changes in absorptivity and/or fluorescence quantum yield of the single molecule. By using a pulsed laser and analyzing the delay times between fluorescence photons and their corresponding excitation pulses, the excited-state lifetime can be determined. Finally, fluorescence spectra, which provide essential information about electronic and vibronic energy structure of the system, are recorded by inserting a beam splitter into the fluorescence emission path. A fraction (∼30%) of the emitted photons can be spectrally dispersed and detected on an EMCCD, while the rest are used for the position estimation required for trapping and recording of fluorescence intensity and lifetime. Correlated changes between these three variables on individual proteins provide particularly useful information to study the photodynamics of these systems.
Analysis of photodegradation pathways: allophycocyanin
Single-molecule experiments enable studies of multi-step processes where each step cannot be precisely synchronized. Indeed, it is often observed that when individuals are directly measured, discontinuous step-like changes in emission brightness are observed. One such process is photodegradation, where individual proteins can transition to multiple intermediate states in response to light before irreversibly entering a dark state (photobleaching).39 Monitoring these processes accesses two important types of information: (1) the dynamics and intermediates in photodegradation; and (2) isolation of individual subunits. In photosynthetic systems, multiple pigments, each in its own local environment, produce overlapping spectroscopic signatures. During the photodegradation process, the photo-induced destruction of each pigment removes its spectroscopic contribution. In the case where the photo-destroyed pigment does not quench neighboring pigments, the laser can serve as a spectroscopic knock-out of individual pigments, reducing the number of pigments contributing to the measurements.Allophycocyanin (APC) is a water-soluble pigment–protein complex located at the base of the primary antenna, the phycobilisome, in cyanobacteria and red algae.40 It is positioned in the middle of the energy transfer chain as the excitation migrates from the antenna complexes towards the reaction center. APC is a trimer with three-fold symmetry, with three weakly-coupled dimers of phycocyanobilin pigments. The trimeric architecture of APC is not stable in solution but can be maintained using chemical crosslinking (XL). Energy relaxes within the dimer in several hundred femtoseconds, and then between the dimers on ∼100 ps timescale.41,42 APC is water soluble, and thus is particularly convenient to study in solution. For this reason, APC was chosen for the first ABEL trap study of the photodynamics of a photosynthetic antenna protein.32
Intensity and lifetime traces of two single XL–APC complexes are shown in Fig. 3a. Single XL–APC complexes exhibit clear stepwise decreases in intensity (left axis). The lifetime (right axis) often, but not always, shows changes correlated with the intensity changes. The correlation between intensity and lifetime is displayed by plotting each intensity level with its concomitant lifetime on a 2D scatter plot in Fig. 3b. The different states of XL–APC can be determined by clustering the points in the scatter plot, as indicated by the colors.32
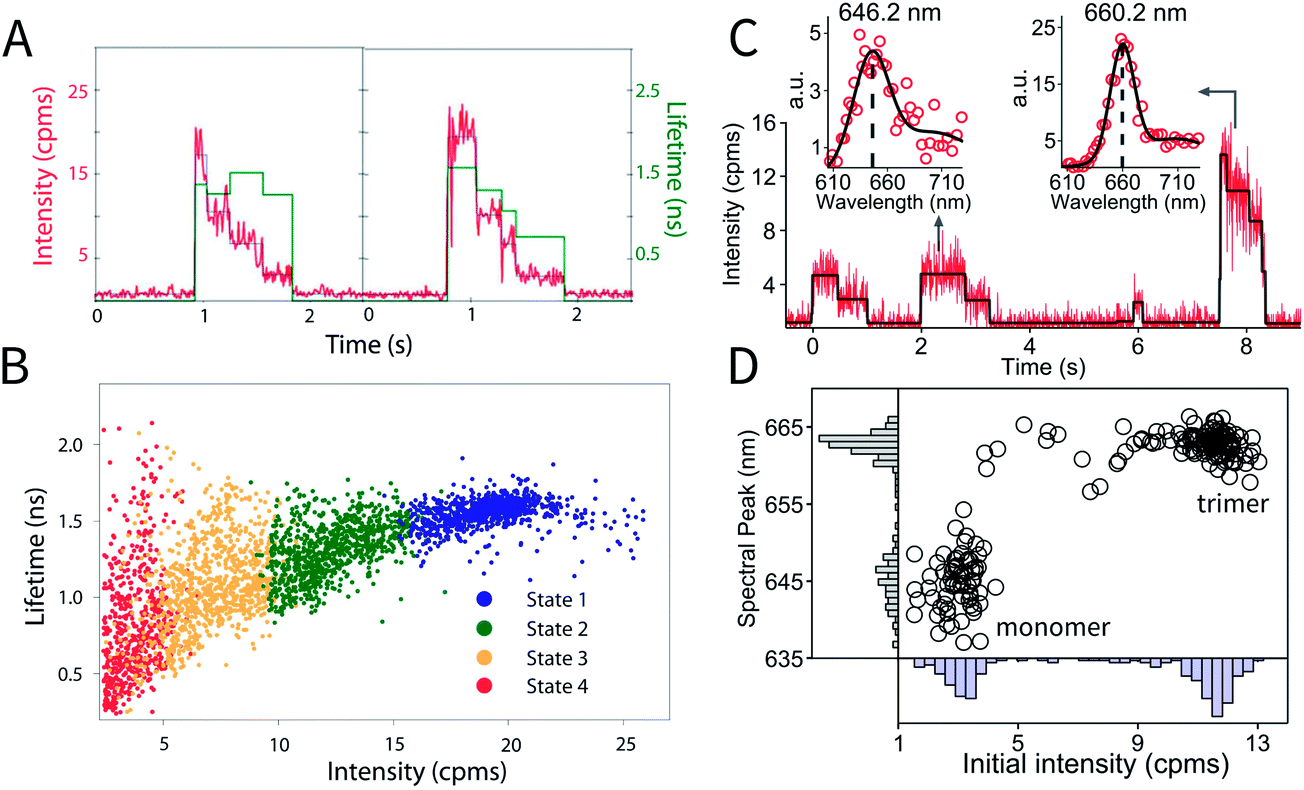 | ||
| Fig. 3 Photodynamics of single APC complexes. (a) Individual XL–APC trimeric complexes exhibit stepwise intensity decreases (red, left axis), as well as both correlated and uncorrelated changes in fluorescence lifetime (green, right axis). (b) The distribution of intensity levels with their concomitant lifetimes is shown on the 2D scatter plot. This distribution can be clustered into states, as shown by the colors in the 2D plot. These figures are reproduced with permission from ref. 32. (c) Intensity and fluorescence emission spectra of individual molecules in a monomer–trimer mixture, following natural dissociation of wild-type APCs. Spectra are fitted with a sum of two Gaussians (solid black curve), with a monomer example shown on the left and trimer on the right. (d) Mapping of initial intensity and spectral peak position of probed wild-type APCs analyzed as in part (c). Two clusters representing monomer and trimer can be identified. | ||
In vivo, the trimeric structure of APC is formed by protein–protein interactions between three monomers in an equilateral triangular geometry. Each monomer protein carries two phycocyanobilin pigments. In the case of APC, in addition to enhancing the light harvesting capability of the complex, trimerization also introduces a ∼30 nm red shift in absorption and ∼20 nm red shift in emission spectrum. Taking advantage of these properties, monomer APCs can be distinguished from trimers in the ABEL trap by analyzing the initial brightness and emission spectrum of each probed molecule (Fig. 3c and d), thus following natural dissociation of wild-type APC at single-molecule concentrations. Here, the first two molecules in Fig. 3c are identified as monomers due to the low initial intensity and blue-shifted emission spectrum peaked at ∼646 nm. The last molecule, identified as an undissociated trimer, emits photons peaked at ∼660 nm and when entering the trap, is ∼3 times brighter compared to the monomers. Moreover, identified monomers frequently show sequential photobleaching of its two pigments (Fig. 3c, 0–1 seconds and 2–3.2 seconds), demonstrating the resolution of a single pigment in the complex. Full details of monomer behavior are currently under study, which could provide valuable insight in the role of self-aggregation in shaping the light-harvesting function of APC.
This experiment identifies the intermediates and timescales of photodegradation of APC. The correlated decreases in fluorescence intensity and lifetime of the trimer indicate an increase in quenching. Because the quenching occurs on a nanosecond timescale (based on the reduction in fluorescence), the energy transfer from APC to other complexes, which occurs much more rapidly, will not be affected. The quenching, therefore, does not interfere with the light-harvesting apparatus. Importantly, the data shows that APC maintains its functionality, during the photodegradation process. As illustrated here, studying proteins on the single molecule level provides an avenue to characterize the photodegradation process, and more generally, multi-step processes, in which each step cannot be synchronized.
Characterization of heterogeneity: peridinin–chlorophyll protein
Photosynthetic proteins exhibit intrinsic heterogeneity and can also fluctuate between conformations at physiological temperatures. Because solution-phase ABEL trap measurements remove any contribution from potentially perturbative immobilization schemes, which may introduce artificial heterogeneity, the intrinsic static and dynamic heterogeneity of a protein can be accurately characterized.Peridinin–chlorophyll protein (PCP) is a water-soluble pigment–protein complex from dinoflagellates. It has a trimeric structure. Each monomer exhibits pseudo-twofold symmetry, containing two Chl, each of which is surrounded by four peridinins that rapidly transfer energy to the enclosed Chl.43 Energy transfers from the Chl to membrane bound complexes to reach the reaction center. As with APC, PCP is a water-soluble protein, and thus it is particularly important to study in an aqueous environment. Although trimeric in vivo, at the low concentrations required for single-molecule studies, ∼99% of the protein is in the monomeric form. The remaining trimers can be identified spectroscopically, as they appear at triple the brightness of the monomeric protein.
In Fig. 4a, fluorescence intensity and lifetime traces for two individual PCP complexes at two different powers are shown. Corresponding spectral dynamics were also recorded by dividing the fluorescence emission into two channels (<680 nm, blue and >680 nm, red, with total fluorescence intensity in black). Fluorescence intensity changes are generally correlated with lifetime changes, but uncorrelated with spectral changes.33
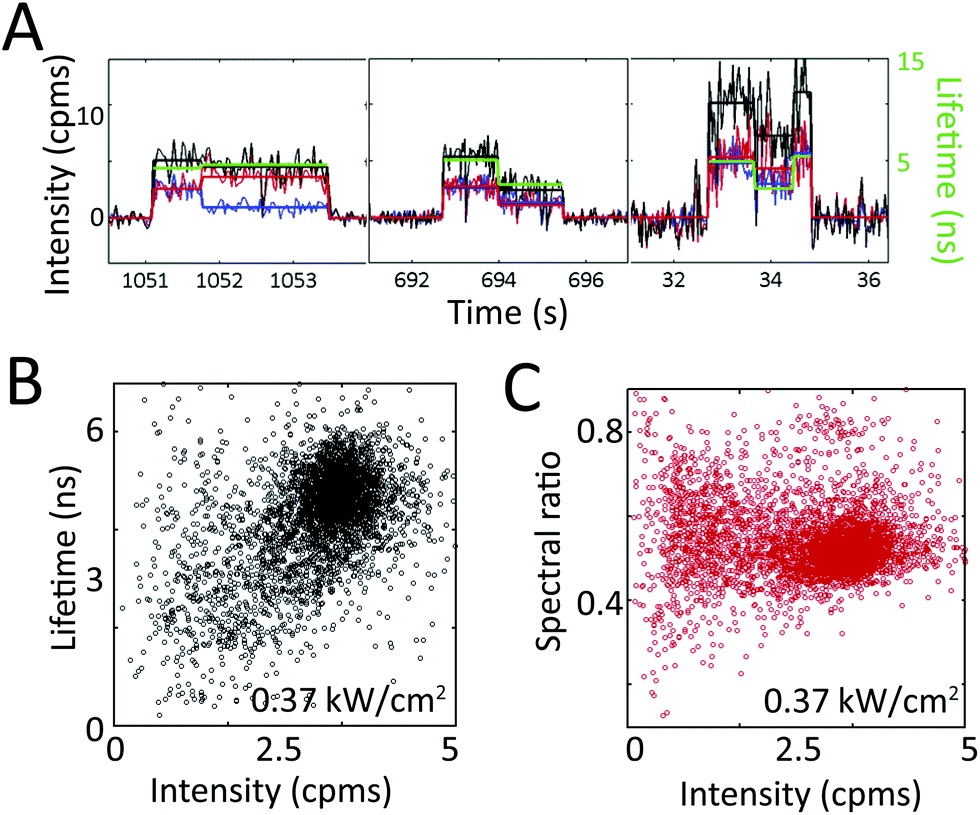 | ||
| Fig. 4 Extracting heterogeneity from single PCP complexes. (a) Single PCP complexes exhibit intensity, lifetime, and spectral dynamics at irradiances of 0.03 (left, center panels) and 0.37 (right panel) kW cm−2. The intensity (black, left axis) is split into two spectral channels (red and blue) at 680 nm, and plotted with its corresponding lifetime (green, right axis). Intensity and lifetime exhibit correlated changes, whereas the two spectral channels occasionally exhibit anti-correlated changes (left panel), indicating a spectral shift. (b) The distribution of total intensity levels with their concomitant lifetimes is shown on the 2D scatter plot. The approximately diagonal elongation indicates correlation between these two variables. (c) The distribution of total intensity levels with their concomitant spectral ratio shows no correlation between these two variables. Reprinted with permission from ref. 33. Copyright 2013 American Chemical Society. | ||
The correlations between intensity, lifetime, and spectral emission are shown in the scatter plots in Fig. 4b and c. Periods of constant intensity are plotted with the concomitant lifetime (Fig. 4b) and spectral ratio (Fig. 4c).33 The diagonal elongation of the distribution in Fig. 4b shows that fluorescence intensity and lifetime are correlated. In contrast, the horizontal elongation of the distribution in Fig. 4c shows that fluorescence intensity and spectrum are uncorrelated, indicating that spectral changes and lifetime changes arise from distinct molecular mechanisms. The difference between these two plots highlights the power of single-molecule techniques. While there is spread in all measurements, we can distinguish multiple different classes of transitions by simultaneously recording multiple variables on single photosynthetic proteins.
From these results, the static and dynamic heterogeneity can be extracted. Individual PCP complexes pumped with 515 nm light at 0.37 kW cm−2 irradiance exhibit fluorescence intensities that range from 1 to 5 cpms (Fig. 4b), lifetimes that range from 0.5 to 6 ns (Fig. 4b), and spectral ratios that range from 0.3 to 0.8 (Fig. 4c), as well as transitions across these ranges (Fig. 4a). The observed heterogeneity and transitions are far larger than what would be expected purely from shot noise. Furthermore, the heterogeneity in these variables increases with excitation intensity.33 As shown here, single-molecule spectroscopy enables the full distribution of variables to be determined, instead of reporting only the average value, as would be available from an ensemble experiment.
Identification of functional forms: light-harvesting complex 2 (LH2)
The behaviors of photosynthetic proteins change in response to light, pH, and temperature. While ensemble structural and functional studies have produced an averaged picture, single-molecule experiments have the ability to identify functional forms of individual complexes, such as asynchronous changes from one state to another, transient intermediates, or rare events.LH2 exhibits an order of magnitude more heterogeneity in excited state energies than many other photosynthetic proteins, and so single-molecule experiments have been particularly insightful. Previous experiments found heterogeneity, but relied on perturbative attachment strategies. In fact, in one paper the authors attribute differences from previous results to differences in attachment strategies.14 With the ABEL trap, we identify the unperturbed functional forms of LH2.
In Fig. 5a, fluorescence intensity and lifetime traces from three sets of experiments on LH2 are shown. These three sets correspond to excitations of each pigment group (carotenoids, B800, and B850) to evaluate their individual contributions. Under all three excitation conditions, these complexes show correlated changes in fluorescence intensity and lifetime. However, at higher intensities, as shown in Fig. 5a, center, asterisk, similar intensities can show dramatically different concomitant lifetimes. In agreement with previous experiments, individual LH2 complexes also showed spectral changes accompanying intensity changes as illustrated in Fig. 5b. However, spectral changes were only observed for ∼3% of the 1073 transitions analyzed.34
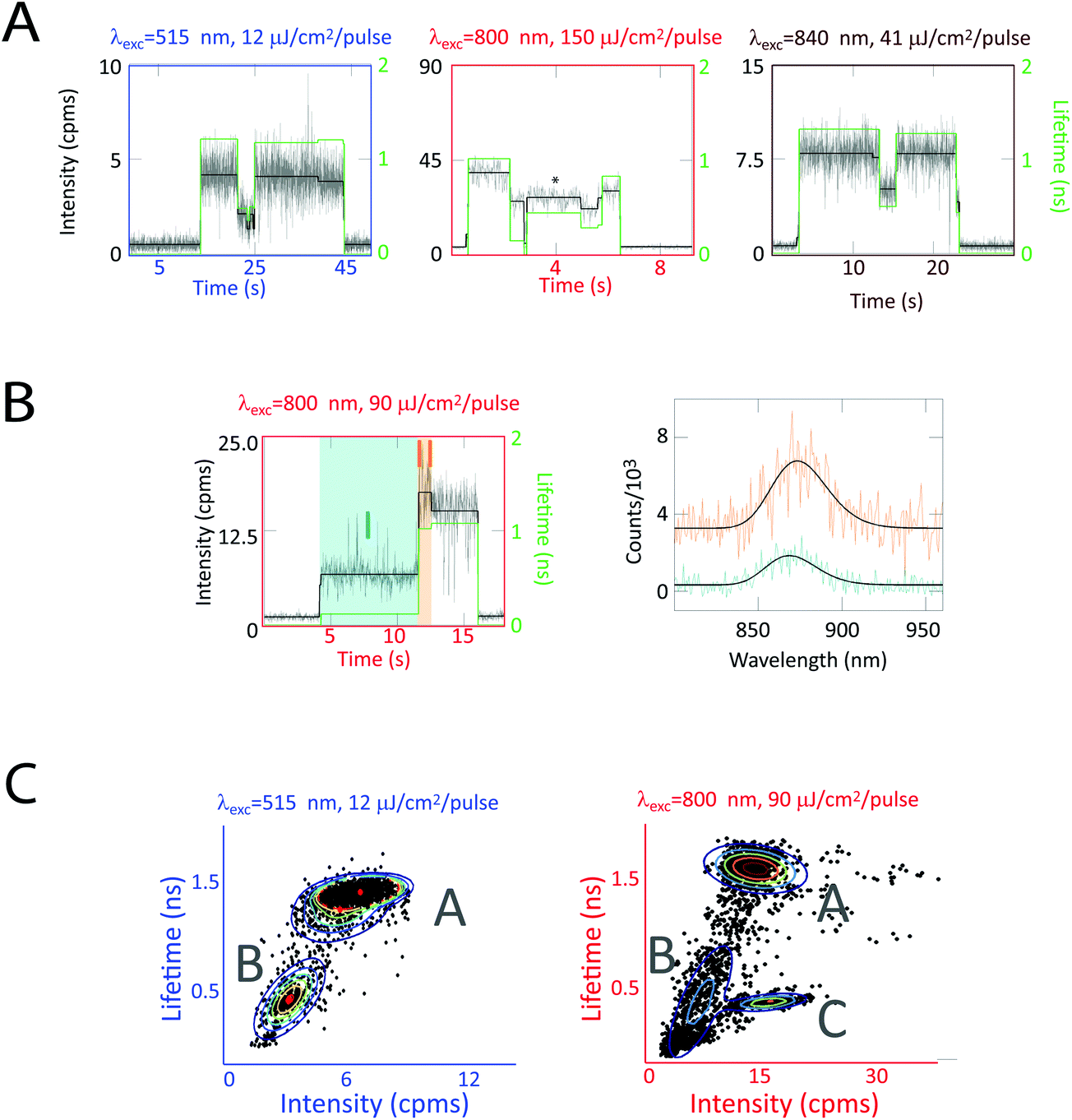 | ||
| Fig. 5 Correlated fluorescence intensity, lifetime, and spectra measurements on single LH2 complexes. (a) Single LH2 complexes exhibit intensity (black, left axis) and lifetime (green, right axis) dynamics for carotenoid (515 nm), B800 (800 nm), and B850 (840 nm) excitation. Correlated intensity–lifetime changes are primarily observed (left, right traces). However, similar intensities can show dramatically different concomitant lifetimes (center trace). (b) Intensity and lifetime changes are occasionally accompanied by spectral changes, as shown by turquoise (868 nm) and orange (873 nm) spectra. (c) 2D intensity–lifetime scatter plots where each period of constant intensity is represented as a dot with its concomitant lifetime. These plots reveal two clusters at low excitation fluence and three at high excitation fluence, as shown by a Gaussian mixture model (rainbow lines) and labeled as states A, B, and C. This figure is reprinted with permission from ref. 34. | ||
Correlations between fluorescence lifetime and intensity reveal three states, labeled as A, B, and C, in the 2D intensity–lifetime plot in Fig. 5c. As shown in the traces in Fig. 5a, individual complexes switch between these three states. Interestingly, all three states exhibit the same spectra within the ∼1 nm resolution of the spectrometer. The intensity–lifetime photodynamics, therefore, are independent of the spectral behaviors previously characterized.34
The intensity dependence of the populations of these three states provides insight into the molecular mechanism behind these states. States A and B appear at all intensities, Fig. 5c, left and right, where state C appears only at high intensities, Fig. 5c, right. The ratio of time in state C increases nonlinearly with excitation fluence. Because of this non-linear dependence combined with molecular modeling, we attribute state C to a photobleached pigment. The rate of transition from state A to state B increased linearly with excitation fluence. Thus, we assign state B to be a local conformational change, triggered by the ∼50–100 kT of an optical excitation. In contrast, the rate of transition from state B to state A is independent of intensity, indicating state B returns to the ground state conformation (state A) via thermal fluctuations.34
State B is a reversible, photoactivated conformation that, notably, has a pathway for quenching excess energy, as shown by the shorter fluorescence lifetimes. This pathway is activated by high excitation intensities and de-activated by thermal motions, thereby providing photoprotection without interfering with function.
Because of the asynchronous nature of the transitions between states A and B, these dynamics would not be accessible in ensemble measurements. Thus, single-molecule spectroscopy, as shown here, serves as a powerful tool to identify functional conformations of photosynthetic complexes.
Conclusions and future outlook
The ABEL trap offers an unprecedented ability to explore the properties of single fluorescent proteins. In a solution-phase environment, the measured heterogeneity and dynamics report on the intrinsic properties of the proteins, instead of on the properties convolved with a perturbation from attachment or encapsulation. As we have described here, this has enabled new insights into the complex structure–function relationships that underlie photosynthetic light harvesting. Specifically, we have analyzed photodegradation pathways, characterized heterogeneity, and identified asynchronous switching between conformations.Current work improving the ABEL trap to enable trapping of dimmer particles will continue to increase the number of photosynthetic proteins that can be studied with this approach. The on-going application of the ABEL trap to study photosynthetic pigment–protein complexes, such as experiments to study higher plants, will offer insight into the variety of protein architectures and dynamics found in photosynthetic organisms.
In future experiments, the impact of the wavelengths of light, pH, temperature, and chemical environment can all be explored. Using the dynamics of single proteins, the ABEL trap has the potential to provide a tool to build up a picture of how photosynthetic systems survive despite the rapid changes in natural conditions. Exploring the microscopic dynamics of single proteins will elucidate the properties of these building blocks that combine to produce the apparatus responsible for fueling most life on earth.
Acknowledgements
We warmly thank the following current and former collaborators: Randall H. Goldsmith, Richard J. Cogdell, and June Southall. We additionally thank Yan Jiang and Hsiang-Yu Yang for helpful discussions. The authors acknowledge the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the US Department of Energy through grant DE-FG02-07ER15892. G.S.S.C. also acknowledges a fellowship from the Stanford Center for Molecular Analysis and Design.References
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell Science, Oxford, 2002 Search PubMed.
- H. Van Amerongen, L. Valkunas and R. Van Grondelle, Photosynthetic excitons, World Scientific Publishing Company Incorporated, Singapore, 2000 Search PubMed.
- A. Ishizaki, T. R. Calhoun, G. S. Schlau-Cohen and G. R. Fleming, Phys. Chem. Chem. Phys., 2010, 12, 7319–7337 RSC.
- Q. Wang, R. H. Goldsmith, Y. Jiang, S. D. Bockenhauer and W. E. Moerner, Acc. Chem. Res., 2012, 45, 1955–1964 CrossRef CAS PubMed.
- A. M. van Oijen, M. Ketelaars, J. Kohler, T. J. Aartsma and J. Schmidt, Science, 1999, 285, 400–402 CrossRef CAS.
- M. A. Bopp, Y. Jia, L. Li, R. J. Cogdell and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10630–10635 CrossRef CAS.
- R. J. Cogdell, A. Gall and J. Köhler, Q. Rev. Biophys., 2006, 39, 227–324 CrossRef CAS PubMed.
- D. Rutkauskas, V. Novoderezhkin, R. J. Cogdell and R. van Grondelle, Biophys. J., 2005, 88, 422–435 CrossRef CAS PubMed.
- S. Wörmke, S. Mackowski, T. Brotosudarmo, C. Jung, A. Zumbusch, M. Ehrl, H. Scheer, E. Hofmann, R. G. Hiller and C. Bräuchle, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 956–964 CrossRef PubMed.
- D. Loos, M. Cotlet, F. C. De Schryver, S. Habuchi and J. Hofkens, Biophys. J., 2004, 87, 2598–2608 CrossRef CAS PubMed.
- L. Ying and X. S. Xie, J. Phys. Chem. B, 1998, 102, 10399–10409 CrossRef CAS.
- G. McDermott, S. Prince, A. Freer, A. Hawthornthwaite-Lawless, M. Papiz, R. Cogdell and N. Isaacs, Nature, 1995, 374, 517–521 CrossRef CAS.
- M. A. Bopp, A. Sytnik, T. D. Howard, R. J. Cogdell and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11271–11276 CrossRef CAS.
- V. I. Novoderezhkin, D. Rutkauskas and R. Van Grondelle, Biophys. J., 2006, 90, 2890–2902 CrossRef CAS PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. Gunner, W. Junge, D. M. Kramer and A. Melis, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- V. Sundström, T. Pullerits and R. van Grondelle, J. Phys. Chem. B, 1999, 103, 2327–2346 CrossRef.
- S. Tubasum, D. Thomsson, R. Cogdell, I. Scheblykin and T. Pullerits, Photosynth. Res., 2012, 111, 41–45 CrossRef CAS PubMed.
- S. Tubasum, R. J. Cogdell, I. G. Scheblykin and T. Pullerits, J. Phys. Chem. B, 2011, 115, 4963–4970 CrossRef CAS PubMed.
- D. Rutkauskas, V. Novoderezkhin, R. J. Cogdell and R. van Grondelle, Biochemistry, 2004, 43, 4431–4438 CrossRef CAS PubMed.
- T. P. Krüger, V. I. Novoderezhkin, C. Ilioaia and R. Van Grondelle, Biophys. J., 2010, 98, 3093–3101 CrossRef PubMed.
- R. Hildner, D. Brinks, J. B. Nieder, R. J. Cogdell and N. F. van Hulst, Science, 2013, 340, 1448–1451 CrossRef CAS PubMed.
- A. Ishizaki and G. R. Fleming, J. Phys. Chem. B, 2011, 115, 6227–6233 CrossRef CAS PubMed.
- V. I. Novoderezhkin, E. Romero, J. P. Dekker and R. van Grondelle, ChemPhysChem, 2011, 12, 681–688 CrossRef CAS PubMed.
- Z. Y. Zhuang, A. I. Jewett, P. Soto and J. E. Shea, Phys. Biol., 2009, 6, 015004 CrossRef PubMed.
- I. Rasnik, S. A. McKinney and T. Ha, Acc. Chem. Res., 2005, 38, 542–548 CrossRef CAS PubMed.
- H. Bai, J. E. Kath, F. M. Zörgiebel, M. Sun, P. Ghosh, G. F. Hatfull, N. D. Grindley and J. F. Marko, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16546–16551 CrossRef CAS PubMed.
- A. E. Cohen and W. E. Moerner, Appl. Phys. Lett., 2005, 86, 093109 CrossRef PubMed.
- A. E. Cohen and W. E. Moerner, Opt. Express, 2008, 16, 6941–6956 CrossRef CAS.
- Q. Wang and W. E. Moerner, ACS Nano, 2011, 5, 5792–5799 CrossRef CAS PubMed.
- Q. Wang and W. E. Moerner, J. Phys. Chem. B, 2012, 117, 4641–4648 CrossRef PubMed.
- A. P. Fields and A. E. Cohen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8937–8942 CrossRef CAS PubMed.
- R. H. Goldsmith and W. E. Moerner, Nat. Chem., 2010, 2, 179–186 CrossRef CAS PubMed.
- S. Bockenhauer and W. E. Moerner, J. Phys. Chem. A, 2013, 117, 8399–8406 CrossRef CAS PubMed.
- G. S. Schlau-Cohen, Q. Wang, J. Southall, R. J. Cogdell and W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10899–10903 CrossRef CAS PubMed.
- P. C. Hiemenz and R. Rajagopalan, Principles of Colloid and Surface Chemistry, revised and expanded, Marcel Dekker, New York, 1997 Search PubMed.
- R. Rigler and E. Elson, Fluorescence Correlation Spectroscopy, Springer, Berlin, 2001 Search PubMed.
- B. Huang, T. D. Perroud and R. N. Zare, ChemPhysChem, 2004, 5, 1523–1531 CrossRef CAS PubMed.
- Q. Wang and W. E. Moerner, Appl. Phys. B, 2010, 99, 23–30 CrossRef CAS PubMed.
- J. Widengren and R. Rigler, Bioimaging, 1996, 4, 149–157 CAS.
- K. Brejc, R. Ficner, R. Huber and S. Steinbacher, J. Mol. Biol., 1995, 249, 424–440 CrossRef CAS PubMed.
- W. F. Beck and K. Sauer, J. Phys. Chem., 1992, 96, 4658–4666 CrossRef CAS.
- J. M. Womick and A. M. Moran, J. Phys. Chem. B, 2011, 115, 1347–1356 CrossRef CAS PubMed.
- E. Hofmann, P. M. Wrench, F. P. Sharples, R. G. Hiller, W. Welte and K. Diederichs, Science, 1997, 272, 1788 Search PubMed.
Footnote |
| † Current address: 332 F St. NE, Apt. B, Washington, DC 20002, USA. |
| This journal is © The Royal Society of Chemistry 2014 |