 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application
Robert
Trondl†
a,
Petra
Heffeter†
bd,
Christian R.
Kowol
ac,
Michael A.
Jakupec
ac,
Walter
Berger
bd and
Bernhard K.
Keppler
*ac
aUniversity of Vienna, Institute of Inorganic Chemistry, Waehringer Str. 42, 1090 Vienna, Austria. E-mail: berhard.keppler@univie.ac.at; Tel: +43 1 4277 52600
bMedical University of Vienna, Institute of Cancer Research and Comprehensive Cancer Center, Borschkegasse 8a, 1090 Vienna, Austria
cUniversity of Vienna, Research Platform Translational Cancer Therapy Research, Waehringer Str. 42, 1090 Vienna, Austria
dMedical University of Vienna, Research Platform Translational Cancer Therapy Research, Borschkegasse 8a, 1090 Vienna, Austria
First published on 4th April 2014
Abstract
NKP-1339 is the first-in-class ruthenium-based anticancer drug in clinical development against solid cancer and has recently been studied successfully in a phase I clinical trial. Ruthenium compounds such as KP1019 (indazolium trans-[tetrachloridobis(1H-indazole)ruthenate(III)]) and NKP-1339 (the sodium salt analogue of KP1019, sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)]) have a high tumour targeting potential based (1) on their strong binding to serum proteins such as albumin and transferrin as well as (2) on their activation in the reductive tumour milieu. The redox activity of ruthenium compounds is believed to represent one major mode of action leading to disturbance of the cellular redox balance and, consequently, induction of G2/M cell cycle arrest, blockage of DNA synthesis, and induction of apoptosis via the mitochondrial pathway. Moreover, potent synergistic activities of NKP-1339 with the clinically approved tyrosine kinase inhibitor sorafenib were recently reported in vitro and in vivo. Taken together, KP1019 and NKP-1339 are promising drug candidates, and especially the very limited side effects observed so far in clinical phase I trials seem to be a major advantage of this class of ruthenium drugs as compared to other chemotherapeutics and targeted anticancer compounds.
A genealogy of antitumour ruthenium compounds
The idea of studying ruthenium compounds as antitumour agents was originally inspired by the observation that certain ruthenium complexes preferentially localize in tumour tissue (reviewed in ref. 4). Additionally, fac-[RuIIICl3(NH3)3] and [RuIICl2(DMSO)4] were found to force the filamentous growth of Escherichia coli by inhibition of cell division – an unusual test model from today's perspective but frequently used at that time.1,2 Subsequent investigations on a panel of ruthenium complexes with different numbers of ammine ligands revealed fac-[RuIIICl3(NH3)3] as most efficacious against EMT-6 sarcoma in mice.3,4 However, fac-[RuIIICl3(NH3)3] was considered unsuitable for further clinical evaluation due to its poor aqueous solubility.5 Consequently, subsequent efforts focused on ionic complexes (in particular anionic species with a higher number of halide ligands) with better solubility (Fig. 1). | ||
| Fig. 1 Genealogy of tumour-inhibiting ruthenium complexes. References refer to the first published evidence for therapeutic activity of the compounds in an in vivo tumour model.5,6,8,15,16 Both NAMI-A and NKP-1339 are currently subject of clinical evaluation. | ||
A lead structure was found in the imidazole-containing complex ICR (KP418), imidazolium trans-[tetrachloridobis(1H-imidazole)ruthenate(III)], which proved therapeutic activity against murine P388 leukaemia and B16 melanoma.6
Moreover, KP418 caused a significant reduction of tumour burden in rats with autochthonous, chemically induced colorectal cancer,7 a model which closely resembles colorectal cancer in patients regarding histopathology, malignant progression, and chemosensitivity patterns. Subsequent studies on a large number of analogues led to the discovery of the indazole analogue KP1019 (indazolium trans-[tetrachloridobis(1H-indazole)ruthenate(III)]), which was found to be superior in its activity against the rat colon cancer model. In these studies, KP1019 treatment yielded efficacy with up to 95% reduction of tumour volume without any mortality (0%) and without any considerable weight loss (6%). In addition, it was found to be superior to 5-fluorouracil, the standard agent against colorectal cancer.8 Based on this promising activity, KP1019 was selected for further (pre)clinical evaluations.
Besides inspiring the development of KP1019, KP418 had also a decisive impact on the field of DMSO-containing ruthenium complexes (pursued by Sava and co-workers), resulting in the synthesis of imidazolium trans-[tetrachlorido(1H-imidazole)(S-dimethyl sulfoxide)ruthenate(III)], NAMI-A (reviewed in ref. 9). Notably, this ruthenium drug turned out, in contrast to KP1019, to affect the process of metastasising rather than acting against the primary tumour or already established metastases. The antimetastatic activity of NAMI-A is probably based on enhanced cell adhesion, integrin-dependent inhibition of cancer cell motility and invasiveness10,11 as well as on inhibition of neoangiogenesis in the tumour tissue.12 Thus, NAMI-A and KP1019, despite their chemical relatedness, behave quite differently in vitro and in vivo.13 Accordingly, they have been clinically developed with completely different scopes and aims.
The most recent representative of this class of compounds is NKP-1339 (sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)], KP1339), the sodium salt analogue of KP1019. This ruthenium compound has originally been prepared as a precursor in the formulation of KP1019 for clinical testing.14 Based on its higher water solubility, NKP-1339 has now been selected as lead candidate for further clinical development. This decision not only facilitated manufacturing and handling for the clinical trials, but also allowed clinical application of larger drug doses to the patients. As many of the early findings relevant for the development of NKP-1339 were made with KP1019, the following sections will deal with both compounds.
Drug delivery through plasma proteins
KP1019 and NKP-1339 are administered intravenously. Thus, the interaction of the complexes with serum proteins is of great relevance. Several studies have shown a strong affinity of KP1019 and NKP-1339 to proteins in the bloodstream, in particular to albumin and transferrin.17,18 Accordingly, it has been suggested that these two proteins act as transport and delivery systems for both ruthenium complexes and, thus, are essential for their tumour targeting. In general, transferrin as well as albumin are considered as highly interesting carriers for drug delivery.19 However, the underlying mechanisms are different. The interest in the iron transport protein transferrin is based on the higher demand of tumours for iron resulting in the overexpression of the transferrin receptor (CD71). Consequently, it has been shown that tumour cells can be specifically targeted by drug binding to transferrin.18,19 Recently, two detailed SEC-ICP/MS analysis studies on the protein-binding pattern of KP1019 in cell culture medium containing 10% fetal calf serum showed that the Ru complex is mainly bound to the albumin- and transferrin-containing protein fraction of 60–80 kDa24,25 (Fig. 2a). This is in accordance with earlier studies on a plasma sample of a KP1019-treated patient, where ruthenium was also exclusively found in this fraction.18 The detailed investigation of the binding of the ruthenium(III) drugs to transferrin was the aim of several in vitro studies. Such, the binding of KP1019 and apolactotransferrin was proven by X-ray crystal structure analysis, revealing that after binding to histidine-253 the two indazole ligands still remain bound to the ruthenium center.26 Moreover, it was shown that the reaction of transferrin with KP1019 or NKP-1339 is very fast and completed within several minutes.27 In contrast, the formation of a transferrin adduct of the imidazole analogue KP418 takes several hours.28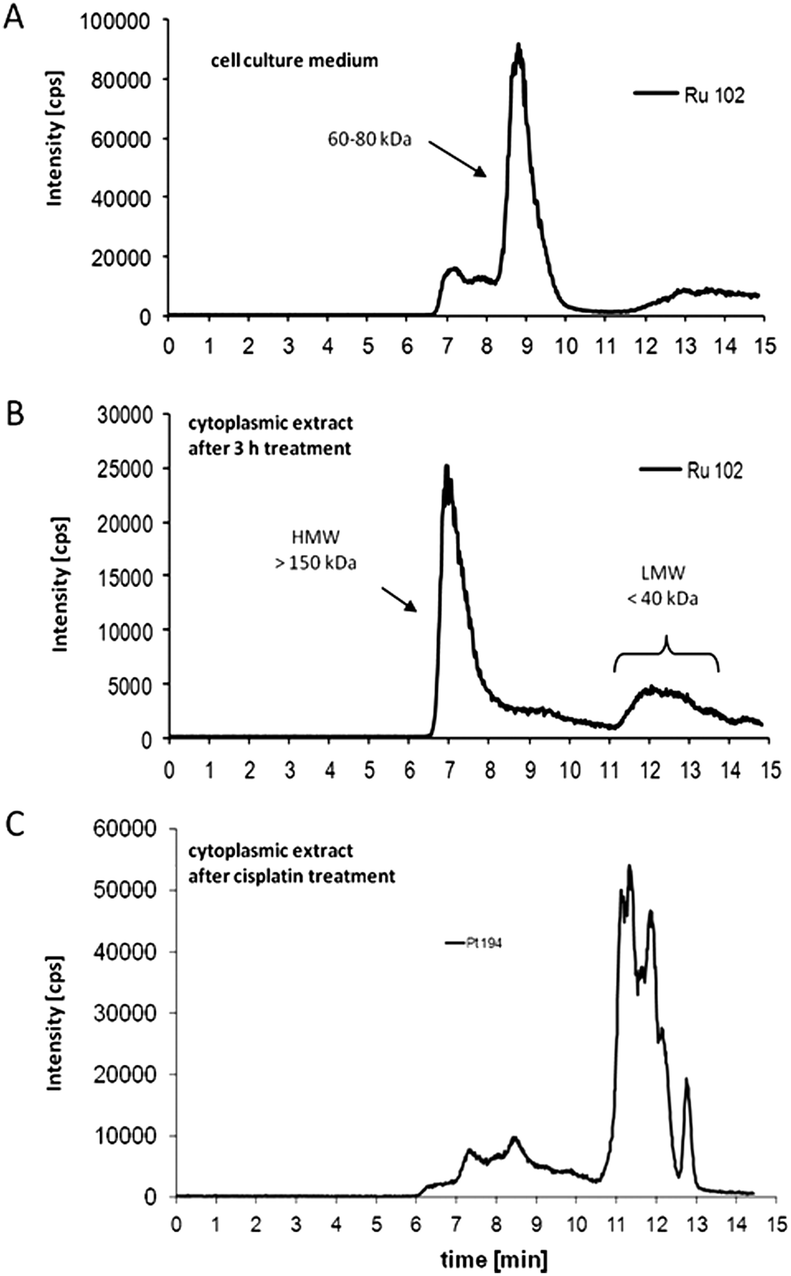 | ||
| Fig. 2 Drug–protein binding patterns of KP1019. (A) Size-exclusion chromatography (SEC)-ICP-MS determination of ruthenium in cell culture medium (supplemented with foetal calf serum) after 3 h of incubation with KP1019. (B) Cytosolic fractions of KP1019-treated KB-3-1 cells were isolated after 3 h treatment, and protein-bound ruthenium was determined by SEC-ICP-MS. (C) Cytosolic fractions of cisplatin-treated KB-3-1 cells were isolated after 3 h treatment with 50 μM cisplatin, and protein-bound platinum was determined by SEC-ICP-MS.24 | ||
Cellular accumulation studies on human cancer cell lines are in good agreement with these data, showing a 10-fold lower uptake of KP418 than of KP1019.29 Additionally, it was shown that the cellular uptake of KP1019 and NKP-1339 is very rapid and finished within the first hour of drug exposure.24 Furthermore, experiments with KP1019–Fe(III)–transferrin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3:1) conjugates showed a >2.5-fold higher uptake of the ruthenium complex in the colon cancer cell line SW480 than after exposure to KP1019 alone. These experiments suggested that a certain amount of iron loading might be needed for optimal transferrin-mediated transport.30
:0.3:1) conjugates showed a >2.5-fold higher uptake of the ruthenium complex in the colon cancer cell line SW480 than after exposure to KP1019 alone. These experiments suggested that a certain amount of iron loading might be needed for optimal transferrin-mediated transport.30
As a consequence of the experimental focus on the transferrin-mediated uptake of KP1019, the “Trojan Horse” hypothesis was developed. It describes a selective delivery of KP1019 into the malignant tissue via transferrin followed by cellular uptake via transferrin receptors. The receptor-mediated incorporation of transferrin results in the formation of endosomes, in which pH is lower (∼5.5) than in the extracellular space (∼7.4), and the drop of pH has been, consequently, thought to trigger the release of the ruthenium complex inside the cell.
Although it is widely accepted that the binding of KP1019/NKP-1339 to serum proteins is essential for their tumour targeting properties, recent analytical studies provoked a new discussion about the importance of transferrin in their drug delivery.31 These studies showed that although adduct formation with transferrin is kinetically preferred, thermodynamically more stable adducts are formed with albumin.9 Moreover, cell-free competition studies with equimolar concentrations of albumin and transferrin showed that less than 20% of KP1019 is bound to transferrin over 10 h incubation time. Taking into account that the albumin concentration in the bloodstream is ∼15-fold higher than that of transferrin, the total amount of transferrin-bound KP1019 has to be expected to be less than 2%. In agreement with these assumptions, SEC-ICP/MS analysis of blood samples taken from a patient of the KP1019 clinical phase I trial showed that the ruthenium complex was preferably bound to albumin, while no KP1019 associated to transferrin was detectable.18 Notably, albumin is known to accumulate in malignant and inflamed tissues due to the combination of leaky blood capillaries with the absence/defect of lymphatic drainage – a phenomenon known as “enhanced permeability and retention (EPR) effect”.22 This effect might also explain the observed tumor accumulation of KP1019, which might then be followed by drug release after albumin degradation.23 Discussions are still ongoing about which plasma protein is most relevant for the tumour-targeting effect of KP1019/NKP-1339 and whether the albumin-bound pool can serve as a reservoir for the transferrin-mediated tumour delivery.
The activation-by-reduction hypothesis
Under physiological conditions, ruthenium mainly exists in two oxidation states, Ru(III) and Ru(II). In general, ruthenium complexes of the oxidation state +III are more inert toward ligand exchange reactions than Ru(II).32 Thus, the aquation process (replacement of chlorido ligands by aqua ligands) is considerably accelerated upon reduction, resulting in “activated” species with higher reactivity toward biomolecules. This behaviour together with data derived from the analysis of pentaammineruthenium(III) complexes prompted Clarke to propose what became known as the “activation-by-reduction” hypothesis.32 This concept is build on the idea that Ru(III) complexes may serve as prodrugs that are activated preferentially in the less oxygenated environment of solid tumours, while sparing normal tissues from toxic effects.33 This is based on the observation that the reduction of Ru(III) to Ru(II) and the subsequent binding to heterocyclic nitrogen bases was found to be catalyzed by mitochondrial, microsomal and soluble subcellular fractions of rat liver cells in the presence of succinate or NADH.33 Moreover, exposure to air markedly decreases the rate of this reaction, probably due to rapid re-oxidation to Ru(III). Due to the higher reactivity, reduction to Ru(II) accelerates coordination to potential target molecules. This mechanism has been assumed to apply by analogy also to azole-containing Ru(III) complexes such as KP1019.32Notably, the activation process depends on the redox potential of the Ru(III)/Ru(II) oxidation states, which in turn strongly depends on the ligands coordinated to the ruthenium centre. Thus, the choice of ligands with suitable electron donor properties enables the tuning of the redox potential to obtain complexes which are redox active in the biological environment, such as KP1019 and NKP-1339.34,35 In case of NKP-1339, electrochemical investigations in phosphate buffer at pH 7 revealed a quasi-reversible reduction wave at +0.03 V vs. NHE (KP418 = −0.25 V vs. NHE).36 As the physiologically accessible redox potential window ranges from around −0.4 V (NADPH, the strongest physiological reductant37) to +0.8 V vs. NHE (dioxygen, the strongest physiological oxidant; NHE = normal hydrogen electrode),38 this potential suggests a possible activation by reduction in the biological environment (in the cellular environment the redox potential of dioxygen is usually only ∼+0.14 V vs. NHE39). It is important to mention that the ruthenium complexes are not reduced effectively while bound to serum proteins,40,41 and thus activation by reduction takes place only after release from the protein inside the cancer cell. Support for the validity of the “activation-by-reduction” hypothesis for the ruthenium compounds has been provided by studies showing an increased cytotoxicity and DNA binding of KP418 under reduced oxygen pressure.42 Also in case of KP1019, nuclear magnetic resonance (NMR) experiments regarding the reduction of Ru(III) to Ru(II) in the presence of ascorbic acid and glutathione (GSH) indicated a complete reduction under buffered conditions within minutes and within 3.5 h, respectively. Furthermore, an increased reactivity of KP1019 was found upon addition of two equivalents of GSH.36 In cell culture experiments, a distinctly higher activity of KP1019 was observed in the presence of ascorbic acid at concentrations from 50 to 700 μM, which went again hand in hand with improved binding to biomolecules in cell culture as well as in cell-free systems.43 Although potent activity of KP1019/NKP-1339 is observed in vivo against diverse cancer types, these findings also implicate that KP1019 might be especially of interest for malignancies of tissues with high endogenous amounts of ascorbic acid (e.g. the pancreas).44
However, with regard to GSH, it has to be mentioned that incubation of KP1019 with a 10-fold excess of GSH resulted in decreased reactivity.36 Accordingly, induction of a low level GSH increase by preincubation with the GSH precursor N-acetylcysteine (NAC) enhanced, while higher concentrations reduced the cytotoxic activity of KP1019 against cancer cells.44 This could be explained by an inactivation of the complex via direct binding of GSH to the ruthenium centre,45 which indicates also that GSH might play a role in resistance of cancer cells against KP1019 (compare section “Drug resistance”). Thus, appropriate concentrations of the reductive agents seem to be important for the activation of KP1019 and NKP-1339.
The question whether the ruthenium centre is reduced in vivo was recently addressed by X-ray absorption spectroscopy (XAS) analysis of tumour and liver samples from mice bearing subcutaneous tumours (either sarcoma 180 treated with KP1019 or NKP-1339, or SW480 carcinoma treated with NKP-1339). This approach is based on the fact that changes of the oxidation state and exchange of donor atoms around the metal centre are reflected in characteristic shifts of edge energies in XAS spectra, as surveyed with a set of model compounds representing a variety of coordination environments around the ruthenium centre. The observed energy shifts were virtually identical in all in vivo settings, and two different scenarios – average coordination patterns of Ru(III)Cl3N2(O/N) or Ru(II)ClN2(O/N)3 – are in best accordance with the spectra.46 Another XAS study was made, which included hepatoma cells treated with KP1019 under cell culture conditions, but no samples obtained from animal tumour models, thus not shedding light on the fate of the compound in vivo.47
Mechanisms underlying the anticancer activity
Based on the “activation-by-reduction” theory, it is not surprising that the mode of action underlying the anticancer activity of KP1019 and NKP-1339 seems to be tightly linked to their redox chemistry (Fig. 3). As already discussed, the activation by reduction leads to a reactive ruthenium(II) complex, which can react with diverse biomolecules.48–51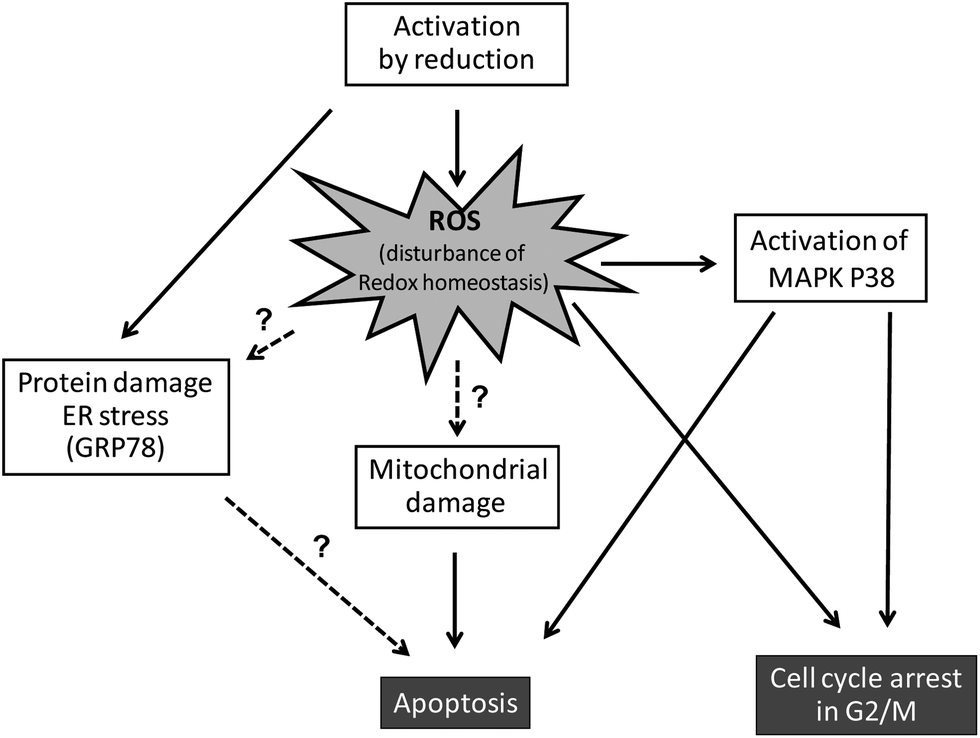 | ||
| Fig. 3 Mechanisms underlying the anticancer activity of KP1019 and NKP-1339. | ||
Although Ru has been detected in nuclei and bound to extracted DNA of cancer cells after drug treatment, there is increasing evidence that the anticancer activity of KP1019 is not primarily based on direct DNA damage.24,32,52 However, in contrast to DNA binding, the reaction of ruthenium compounds with intracellular proteins has been far less investigated. Recent studies on the intracellular protein-binding patterns of KP101924,25 and NKP-133924 in the cytosol of cancer cells showed that Ru-binding can be detected mainly in protein factions of two size classes: protein aggregates/complexes >700 kDa and low molecular weight protein (complexes) <40 kDa. Notably, this intracellular drug binding patterns of KP1019/NKP-1339 strongly differed from those of cells exposed to cisplatin (Fig. 2b and c).
Additionally, the glucose-regulated protein of 78 kDa (GRP78) was recently suggested as one cytosolic target for NKP-1339.53 The endoplasmic reticulum (ER) chaperone GRP78 is a key factor of the unfolded protein response (UPR),54 suggesting an interaction of NKP-1339 with the protein maintenance machinery of the cell. Cancer cells alter various processes to enable high proliferation, increased glucose metabolism, and resistance to cellular stresses and death initiation. One way to compensate cellular stresses is to upregulate key factors of the UPR, such as ER chaperones and UPR target genes, which was also observed in human tumor samples. In contrast to most normal cells, which are not under ER stress and where the UPR is normally in a non-active state, cancer cells seem to be critically dependent on the preservation of their ER folding capacity. Thus, targeting the UPR has been proposed as a promising new strategy for cancer treatment.
Besides direct biomolecule damage/interactions, the redox activity of ruthenium(III) compounds can be expected to interfere with the cellular redox balance via direct as well as indirect mechanisms.55 Such ruthenium(III) compounds might participate in Fenton-like reactions leading to generation of reactive oxygen species (ROS). On the other hand, the reaction of ruthenium(III) drugs with GSH (e.g. by formation of GSH-conjugates45) can be expected to induce depletion of the intracellular GSH pools, which renders cells more susceptible to endogenous and exogenous oxidative stress. Indeed, formation of intracellular ROS was shown in human colon cancer cells after short-time KP1019 treatment.56 Moreover, depletion of the intracellular GSH pools by pre-treatment with buthionine sulfoximine (BSO) led to increased sensitivity of cancer cells to KP1019.57 These findings argue for the participation of ROS in the mechanism underlying anticancer activity of this drug.
Interestingly, a recent X-ray fluorescence imaging study of single cells found cytosolic co-localisation of Ru with iron as well as changes in the intracellular iron distribution.58 This might indicate that, based on its similarities to iron, KP1019 might also interfere with intracellular iron binding proteins and, consequently, iron metabolism. However, this aspect has so far not been investigated in more detail. Interactions with the isolated heme-iron-dependent protein cytochrome c have been reported, but whereas KP1019 was found to induce conformational changes in both apo- and holocytochrome c according to circular dichroism spectra, a possible interference with iron was not addressed here specifically either.59
NKP-1339 (as well as NAMI-A) has recently been identified as a direct nitric oxide (NO˙) scavenger.60 Reaction with NO˙ was shown to result in reduction of Ru(III) and formation of a [Ru(II)–NO+] moiety. As NO˙ is a known messenger for diverse physiological signalling processes, especially in vascular homeostasis and neurotransmission as well as inflammatory/immune response and tumour progression,61 this might suggest some effects of KP1019 and NKP-1339 on endothelial cell migration and angiogenesis.
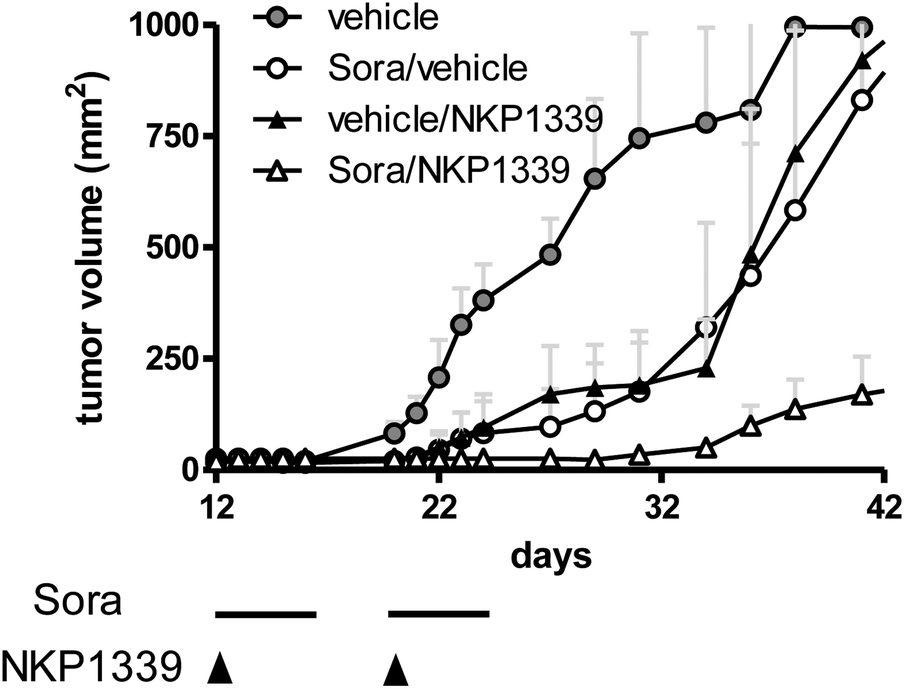 | ||
| Fig. 4 Hep3B xenografts were grown in Balb/c SCID mice and treated with NKP-1339 (30 mg kg−1; i.v.; once a week) and/or sorafenib (25 mg kg−1; p.o.; five consecutive days per week) for two weeks. | ||
As a consequence of this (redox) stress, treatment with KP1019 and NKP-1339 was shown to induce apoptosis of cancer cells via the mitochondrial pathway.52,26 Recent studies indicated that NKP-1339 treatment (comparable to NAMI-A62 but in contrast to KP41863) leads to cell cycle arrest in the G2/M phase.64 In addition, this cell cycle arrest was found to be accompanied by upregulation of the p38 mitogen-activated-protein kinase (MAPK) stress response pathway. The anticancer activity of KP1339 could be further enhanced by combination with the multi-kinase inhibitor sorafenib (BAY 43-9006, Nexavar) (Fig. 4), which targets besides raf serine/threonine kinases also diverse other oncogenic and/or pro-angiogenic receptor tyrosine kinases such as Flt-3, c-kit, RET, VEGFR-1/2/3 and PDGFR-1.64 Notably, synergistic activity with NKP-1339 was found in sorafenib-resistant as well as sorafenib-responsive cell lines and was based on significantly enhanced cellular accumulation of both drugs as well as abrogation of KP1339-induced G2/M arrest by sorafenib. Together these effects resulted in enhanced apoptosis induction and complete loss of mitotic cells in cell culture as well as in a xenograft model in vivo.64
Drug resistance
The occurrence of drug resistance (either intrinsically or acquired during drug treatment) is one of the major obstacles for the treatment of cancer at the disseminated stage. The mechanisms which can lead to resistance are multiple and can involve basically all steps from reduced tumour delivery and cellular drug uptake to defects in apoptosis execution. Besides platinum drugs, the characterisation of relevant resistance mechanisms against metal compounds is still at a very early stage. As already discussed above, GSH is one of the most important intracellular reductants, which is believed to be involved in the activation of ruthenium compounds. Additionally, enhanced GSH levels are frequently associated with resistance against sulfur-affine compounds such as the platinum(II) drugs cis- or oxaliplatin. The impact of enhanced GSH levels on the anticancer activity of ruthenium compounds is still not fully understood. On the one hand, depletion of intracellular GSH levels led to increased sensitivity against KP1019,57 while pre-treatment with high doses of the radical scavenger and precursor of GSH, N-acetylcysteine, protected human colon carcinoma cells from the anticancer activity of KP1019.56 On the other hand, it has to be kept in mind that studies on the cisplatin-resistant cell models O-342/DPP and A2780cis revealed that these cells, which are displaying enhanced GSH levels, are not cross-resistant in vitro and in vivo against several Ru(III) drugs including KP1019.25,65,66 Additionally and in contrast to observations with cisplatin, no reduction of GSH levels was observed in rat kidney cells after treatment of animals with KP1019.67 Together, these results suggest that the anticancer activity of ruthenium compounds might be at least less susceptible to enhanced cellular GSH levels of drug-resistant cancer cells. Overall, the somewhat contradictory findings regarding the impact of GSH on the anticancer activity of these ruthenium compounds might be explained by the opposite impact of activation by reduction and detoxification via conjugation. Nevertheless, there is increasing evidence that KP1019/NKP-1339 treatment leads to a changed GSH/GSSG ratio and disturbances of the intracellular redox balance, which also impacts on the resistance profile of these drugs.45Another well described mechanism which is frequently involved in drug resistance is the overexpression of ATP-driven efflux proteins, especially ABC transporters. The family of ABC transporter proteins is large, but only a few of its family members (such as several ABCC members, ABCB1 and ABCG2) have been associated with drug resistance. We have recently studied the impact of these transport proteins on the anticancer activity of KP1019.68 In this study, it was discovered that ABCC1 (multidrug-resistance protein 1, MRP1) expression had no impact on the anticancer activity of KP1019.68 This is especially of interest as most ABCC family members have a strong substrate affinity for GSH conjugates. In contrast to ABCC1, several ABCB1 (P-glycoprotein, P-gp)-overexpressing cell lines showed weak resistance (about 2-fold) to KP1019 based on reduced drug accumulation, compared to >100-fold resistance to common ABCB1 substrates (e.g. doxorubicin). Notably, the interaction of KP1019 with ABCB1 was strongly influenced by serum protein levels, indicating that the strong binding to serum proteins might protect KP1019 from recognition and subsequent export by ABCB1. This is in accordance with studies with the high affinity ABCB1 substrate doxorubicin, where conjugation to the iron transport protein transferrin was able to circumvent efflux by ABCB1 and, consequently, to enhance cytotoxic activity against ABCB1-overexpressing cells.20,21 Likewise, a nanopreparation of KP1019 was highly effective against ABCB1-overexpressing cells and generally led to enhanced drug uptake via endocytosis.69 To investigate a possible acquired resistance to KP1019, KB-3-1 cells were selected by exposure to stepwise increasing concentrations of KP1019. Notably, this resulted only in a 2-fold resistance to KP1019,68 which was not based on reduced drug accumulation and ABC transporter overexpression. The mechanism underlying KP1019 resistance is still under investigation. Recent studies using comparative genomic hybridization revealed that KP1019-resistant KB-3-1 cells are characterized by several chromosome losses including chromosome 5, 7q, 12, 13q and 16q in comparison to their parental cell line (unpublished results). Moreover, no gene amplification was observed, indicating that the acquired resistance of this cell line is probably supported by gene loss and/or regulated by epigenetic mechanisms.
Evaluation in clinical studies
Both, KP1019 and NKP-1339, belong to the very few ruthenium compounds which have been already evaluated in clinical studies.53,70,71 In fact, the antimetastatic drug NAMI-A is the only other ruthenium compound studied in a phase I trial.72 In the case of KP1019 an open-label flat-dose escalation trial was performed in patients with advanced solid tumours without further therapeutic options.71 Eight patients were accrued receiving intravenous KP1019 doses from 25 to 600 mg twice weekly over three weeks. The pharmacokinetic analysis suggested that, in accordance with previous observations in vitro, the drug is rapidly bound to plasma proteins and has a long half-life (about 69–284 h). Clearance and the volume of distribution were low, and the AUC (area under the curve) and Cmax increased dose-proportionally. Notably, KP1019 was extremely well tolerated with only very limited side effects, and, accordingly, the maximum tolerated dose was not reached (dose escalation had to be stopped because of the high infusion volume required for solubility reasons). This is in contrast to the study evaluating NAMI-A, where painful blister formation at hands, fingers, and toes was considered as the dose-limiting toxicity.72Despite this dose limitation, five of six evaluable patients showed disease stabilization (including one minor remission) for eight to ten weeks. Interestingly, this clinical effect was not strictly depending on the applied dose. Consequently, the study suggested a 400–600 mg flat dose regimen for a phase II study with a longer application time (10 weeks), based on data derived from preclinical animal models and the observation of long-lasting stable diseases.
In order to avoid dose limitation by the high infusion volume, the clinical development was recently redirected to the better soluble NKP-1339, which allows the application of higher doses. The respective phase I study is completed, and the interim data were presented at the ASCO meeting 2012.73 NKP-1339 was administered as 30–90 min infusion (based on volume to be infused) on days 1, 8 and 15 of a 28 day cycle. A total of 34 patients with various solid tumours have been treated so far at 9 different dose levels (20, 40, 80, 160, 320, 420, 500, 625 and 780 mg m−2 body weight). Very minor side effects have been observed. At higher doses a transient green discoloration of the plasma was observed without any clinical symptoms. Unrelated to this phenomenon, only grade 1–2 pyrexia and/or rigors were observed so far in some patients, which were prevented in subsequent patients by steroid-based premedication. Dose limiting toxicity of grade 2–3 nausea, with grade 2 creatinine levels in one patient occurred at 780 mg m−2. Also for NKP-1339, preliminary pharmacokinetic analyses indicated dose proportionality of Cmax and AUC, suggesting linear pharmacokinetics. Partial response was observed in one patient with a neuroendocrine tumour (NET) and stable disease (for up to 88+ weeks) in seven patients, including NET (two patients), colorectal cancer, sarcoma, and unknown primary (one patient each), as well as two patients with non-small cell lung cancer (NSCLC). As also in the phase I trial of NAMI-A the one patient (out of 24) which suffered from NSCLC experienced stable disease,72 these data might indicate enhanced sensitivity of NSCLC to ruthenium compounds. Whether clinical stabilisation/response correlates with any molecular factor possibly influencing the pharmacodynamics of NKP-1339 (e.g., glutathione) can be a worthwhile subject for further investigation.
Conclusions
Research in the field of ruthenium-based anticancer drugs, which steadily progressed over the last 20 years, is now entering the crucial phase towards clinical application. The tremendous efforts on the characterization of the chemical and biological properties of ruthenium-based drugs made clear that ruthenium compounds are a unique class of anticancer compounds. Several key studies elucidated that ruthenium-based complexes exhibit different mechanisms of accumulation, uptake, activation and mode of action within the cell compared to established drugs. Besides the promising anticancer activities in heavily pre-treated patients, especially the very limited adverse effects observed so far in clinical phase I trials of KP1019 and NKP-1339 are a major advantage in comparison to other anticancer metal drugs. Beside low general toxicity, tumour selectivity and the development of predictive biomarkers are the most important criteria for successful clinical approval. Therefore, gaining a deeper insight into the interactions of NKP-1339 with target molecules within the cancer cell will be one of the main challenges for further investigations.Acknowledgements
References
- C. Monti-Bragadin, L. Ramani, L. Samer, G. Mestroni and G. Zassinovich, Antimicrob. Agents Chemother., 1975, 7, 825–827 CrossRef CAS.
- J. R. Durig, J. Danneman, W. D. Behnke and E. E. Mercer, Chem.-Biol. Interact., 1976, 13, 287–294 CrossRef CAS.
- M. J. Clarke, S. Bitler, D. Rennert, M. Buchbinder and A. D. Kelman, J. Inorg. Biochem., 1980, 12, 79–87 CrossRef CAS.
- S. C. Srivastava, L. F. Mausner and M. J. Clarke, Prog. Clin. Biochem. Med., 1989, 10, 111–149 CAS.
- M. J. Clarke, Met. Ions Biol. Syst., 1980, 11, 231–283 CAS.
- B. K. Keppler and W. Rupp, J. Cancer Res. Clin. Oncol., 1986, 111, 166–168 CrossRef CAS.
- F. T. Garzon, M. R. Berger, B. K. Keppler and D. Schmaehl, Cancer Chemother. Pharmacol., 1987, 19, 347–349 CrossRef CAS.
- B. K. Keppler, M. Henn, U. M. Juhl, M. R. Berger, R. Niebl and F. E. Wagner, Prog. Clin. Biochem. Med., 1989, 10, 41–69 CAS.
- I. Bratsos, T. Gianferrara, E. Alessio, C. G. Hartinger, M. A. Jakupec and B. K. Keppler, in Bioinorganic Medicinal Chemistry, Wiley Online Library, 2011, p. 151 Search PubMed.
- G. Sava, F. Frausin, M. Cocchietto, F. Vita, E. Podda, P. Spessotto, A. Furlani, V. Scarcia and G. Zabucchi, Eur. J. Cancer, 2004, 40, 1383–1396 CrossRef CAS PubMed.
- F. Frausin, V. Scarcia, M. Cocchietto, A. Furlani, B. Serli, E. Alessio and G. Sava, J. Pharmacol. Exp. Ther., 2004, 313, 227–233 CrossRef PubMed.
- A. Vacca, M. Bruno, A. Boccarelli, M. Coluccia, D. Ribatti, A. Bergamo, S. Garbisa, L. Sartor and G. Sava, Br. J. Cancer, 2002, 86, 993–998 CrossRef CAS PubMed.
- A. Bergamo, A. Masi, M. A. Jakupec, B. K. Keppler and G. Sava, Met.-Based Drugs, 2009, 2009, 681270 CAS.
- W. Peti, T. Pieper, M. Sommer, B. K. Keppler and G. Giester, Eur. J. Inorg. Chem., 1999, 1999, 1551–1555 CrossRef.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- G. Sava, S. Pacor, G. Mestroni and E. Alessio, Clin. Exp. Metastasis, 1992, 10, 273–280 CrossRef CAS.
- F. Kratz, B. K. Keppler, M. Hartmann, L. Messori and M. R. Berger, Met.-Based Drugs, 1996, 3, 15–23 CrossRef CAS PubMed.
- M. Sulyok, S. Hann, C. G. Hartinger, B. K. Keppler, G. Stingeder and G. Koellensperger, J. Anal. At. Spectrom., 2005, 20, 856 RSC.
- F. Kratz and B. Elsadek, J. Controlled Release, 2011, 161, 1–16 Search PubMed.
- M. Fritzer, K. Barabas, V. Szuts, A. Berczi, T. Szekeres, W. P. Faulk and H. Goldenberg, Int. J. Cancer, 1992, 52, 619–623 CrossRef CAS PubMed.
- M. Fritzer, T. Szekeres, V. Szuets, H. N. Jarayam and H. Goldenberg, Biochem. Pharmacol., 1996, 51, 489–493 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- P. Heffeter, K. Böck, B. Atil, M. A. Reza Hoda, W. Körner, C. Bartel, U. Jungwirth, B. K. Keppler, M. Micksche, W. Berger and G. Koellensperger, JBIC, J. Biol. Inorg. Chem., 2010, 15, 737–748 CrossRef CAS PubMed.
- M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 3, 591–599 RSC.
- C. A. Smith, A. J. Sutherland-Smith, B. K. Keppler, F. Kratz and E. N. Baker, JBIC, J. Biol. Inorg. Chem., 1996, 1, 424–431 CrossRef CAS.
- A. R. Timerbaev, A. V. Rudnev, O. Semenova, C. G. Hartinger and B. K. Keppler, Anal. Biochem., 2005, 341, 326–333 CrossRef CAS PubMed.
- F. Kratz, M. Hartmann, B. Keppler and L. Messori, J. Biol. Chem., 1994, 269, 2581–2588 CAS.
- S. Kapitza, M. Pongratz, M. A. Jakupec, P. Heffeter, W. Berger, L. Lackinger, B. K. Keppler and B. Marian, J. Cancer Res. Clin. Oncol., 2004, 131, 101–110 CrossRef PubMed.
- M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46 RSC.
- A. K. Bytzek, K. Boeck, G. Hermann, S. Hann, B. K. Keppler, C. G. Hartinger and G. Koellensperger, Metallomics, 2011, 3, 1049–1055 RSC.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209–233 CrossRef CAS.
- A. D. Kelman, M. J. Clarke, S. D. Edmonds and H. J. Peresie, J. Clin. Hematol. Oncol., 1977, 7, 274–288 CAS.
- M. A. Jakupec, E. Reisner, A. Eichinger, M. Pongratz, V. B. Arion, M. Galanski, C. G. Hartinger and B. K. Keppler, J. Med. Chem., 2005, 48, 2831–2837 CrossRef CAS PubMed.
- E. Reisner, V. B. Arion, B. K. Keppler and A. J. L. Pombeiro, Inorg. Chim. Acta, 2008, 361, 1569–1583 CrossRef CAS PubMed.
- P. Schluga, C. G. Hartinger, A. Egger, E. Reisner, M. Galanski, M. A. Jakupec and B. K. Keppler, Dalton Trans., 2006, 1796–1802 RSC.
- F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS.
- C. S. Sevier and C. A. Kaiser, Nat. Rev. Mol. Cell Biol., 2002, 3, 836–847 CrossRef CAS PubMed.
- B. Halliwell and J. M. Gutteridge, Free Radicals in Biology and Medicine, Oxford University Press, 4th edn, 2007 Search PubMed.
- F. Piccioli, S. Sabatini, L. Messori, P. Orioli, C. G. Hartinger and B. K. Keppler, J. Inorg. Biochem., 2004, 98, 1135–1142 CrossRef CAS PubMed.
- A. R. Timerbaev, L. S. Foteeva, A. V. Rudnev, J. K. Abramski, K. Połeć-Pawlak, C. G. Hartinger, M. Jarosz and B. K. Keppler, Electrophoresis, 2007, 28, 2235–2240 CrossRef CAS PubMed.
- D. Frasca, J. Ciampa, J. Emerson, R. S. Umans and M. J. Clarke, Met.-Based Drugs, 1996, 3, 197–209 CrossRef CAS PubMed.
- C. Bartel, A. E. Egger, M. A. Jakupec, P. Heffeter, M. Galanski, W. Berger and B. K. Keppler, JBIC, J. Biol. Inorg. Chem., 2011, 16, 1205–1215 CrossRef CAS PubMed.
- S. Brown, M. Georgatos, C. Reifel, J. H. Song, S. H. Shin and M. Hong, Endocrine, 2002, 18, 91–96 CrossRef CAS PubMed.
- D. R. Frasca and M. J. Clarke, J. Am. Chem. Soc., 1999, 121, 8523–8532 CrossRef CAS.
- A. A. Hummer, P. Heffeter, W. Berger, M. Filipits, D. Batchelor, G. E. Büchel, M. A. Jakupec, B. K. Keppler and A. Rompel, J. Med. Chem., 2013, 56, 1182–1196 CrossRef CAS PubMed.
- A. Levina, J. B. Aitken, Y. Y. Gwee, Z. J. Lim, M. Liu, A. M. Singharay, P. F. Wong and P. A. Lay, Chem.–Eur. J., 2013, 19, 3609–3619 CrossRef CAS PubMed.
- M. Groessl, Y. O. Tsybin, C. G. Hartinger, B. K. Keppler and P. J. Dyson, JBIC, J. Biol. Inorg. Chem., 2010, 15, 677–688 CrossRef CAS PubMed.
- A. Küng, T. Pieper, R. Wissiack, E. Rosenberg and B. K. Keppler, JBIC, J. Biol. Inorg. Chem., 2001, 6, 292–299 CrossRef.
- A. Egger, V. B. Arion, E. Reisner, B. Cebrián-Losantos, S. Shova, G. Trettenhahn and B. K. Keppler, Inorg. Chem., 2005, 44, 122–132 CrossRef CAS PubMed.
- S. K. Stevens, A. P. Strehle, R. L. Miller, S. H. Gammons, K. J. Hoffman, J. T. McCarty, M. E. Miller, L. K. Stultz and P. K. Hanson, Mol. Pharmacol., 2012, 83, 225–234 CrossRef PubMed.
- M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511–2533 CrossRef CAS PubMed.
- N. R. Dickson, S. F. Jones, H. A. Burris, R. K. Ramanathan, G. J. Weiss, J. R. Infante, J. C. Bendell, W. McCulloch and D. D. Von Hoff, J. Clin. Oncol., 2011, 29 Search PubMed , ASCO Meeting Supplement, Abstract #2607.
- M. Ni, Y. Zhang and A. S. Lee, Biochem. J., 2011, 434, 181–188 CrossRef CAS PubMed.
- U. Jungwirth, C. R. Kowol, B. K. Keppler, C. G. Hartinger, W. Berger and P. Heffeter, Antioxid. Redox Signaling, 2011, 15, 1085–1127 CrossRef CAS PubMed.
- S. Kapitza, M. A. Jakupec, M. Uhl, B. K. Keppler and B. Marian, Cancer Lett., 2005, 226, 115–121 CrossRef CAS PubMed.
- P. Heffeter, U. Jungwirth, M. Jakupec, C. Hartinger, M. Galanski, L. Elbling, M. Micksche, B. Keppler and W. Berger, Drug Resist. Updates, 2008, 11, 1–16 CrossRef CAS PubMed.
- J. Aitken, S. Antony, C. Weekley and B. Lai, Metallomics, 2012, 4, 1051 RSC.
- L. Trynda-Lemiesz, Acta Biochim. Pol., 2004, 51, 199–205 CAS.
- L. Morbidelli, S. Donnini, S. Filippi, L. Messori, F. Piccioli, P. Orioli, G. Sava and M. Ziche, Br. J. Cancer, 2003, 88, 1484–1491 CrossRef CAS PubMed.
- A. Bencini, P. Failli, B. Valtancoli and D. Bani, Cardiovasc. Hematol. Agents Med. Chem., 2010, 8, 128–146 CrossRef CAS.
- A. Bergamo, S. Zorzet, M. Cocchietto, M. E. Carotenuto, M. Magnarin and G. Sava, Anticancer Res., 2001, 21, 1893–1898 CAS.
- S. Zorzet, A. Bergamo, M. Cocchietto, A. Sorc, B. Gava, E. Alessio, E. Iengo and G. Sava, J. Pharmacol. Exp. Ther., 2000, 295, 927–933 CAS.
- P. Heffeter, B. Atil, K. Kryeziu, D. Groza, G. Koellensperger, W. Körner, U. Jungwirth, T. Mohr, B. K. Keppler and W. Berger, Eur. J. Cancer, 2013, 49, 3366–3375 CrossRef CAS PubMed.
- S. Fruehauf and W. J. Zeller, Cancer Res., 1991, 51, 2943–2948 CAS.
- S. Fruehauf and W. J. Zeller, Cancer Chemother. Pharmacol., 1991, 27, 301–307 CrossRef CAS.
- L. Kersten, H. Bräunlich, B. K. Keppler, C. Gliesing, M. Wendelin and J. Westphal, J. Appl. Toxicol., 1998, 18, 93–101 CrossRef CAS.
- P. Heffeter, M. Pongratz, E. Steiner, P. Chiba, M. A. Jakupec, L. Elbling, B. Marian, W. Körner, F. Sevelda, M. Micksche, B. K. Keppler and W. Berger, J. Pharmacol. Exp. Ther., 2004, 312, 281–289 CrossRef PubMed.
- P. Heffeter, A. Riabtseva, Y. Senkiv, C. Kowol, W. Körner, U. Jungwith, N. Mitina, B. K. Keppler, T. Konstantinova, I. Yanchuk, R. Stoika, A. Zaichenko and W. Berger, J. Biomed. Nanotechnol., 2014, 10, 877–884 CrossRef CAS PubMed.
- F. Lentz, A. Drescher, A. Lindauer, M. Henke, R. A. Hilger, C. G. Hartinger, M. E. Scheulen, C. Dittrich, B. K. Keppler and U. Jaehde, Anti-Cancer Drugs, 2009, 20, 97–103 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- J. M. Rademaker-Lakhai, D. B. Van Desiree, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- D. S. Thompson, G. J. Weiss, S. F. Jones, H. A. Burris, R. K. Ramanathan, J. R. Infante, J. C. Bendell, A. Ogden and D. D. Von Hoff, J. Clin. Oncol., 2012, 30 CAS , ASCO Meeting Supplement, Abstract# 3033.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |