 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Si–H addition followed by C–H bond activation induced by a terminal thorium imido metallocene: a combined experimental and computational study†
Wenshan
Ren
ab,
Enwei
Zhou
a,
Bo
Fang
a,
Guofu
Zi
*a,
De-Cai
Fang
*a and
Marc D.
Walter
*c
aDepartment of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: gzi@bnu.edu.cn; dcfang@bnu.edu.cn; Fax: +86-10-58802075; Tel: +86-10-58806051
bCollege of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, China
cInstitut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: mwalter@tu-bs.de; Fax: +49-531-3915387; Tel: +49-531-3915312
First published on 9th June 2014
Abstract
The Si–H bond addition to a terminal actinide imido complex was comprehensively studied. The base-free thorium imido [η5-1,2,4-(Me3C)3C5H2]2Th![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(p-tolyl) (1) activates Si–H bonds in PhSiH3 or Ph2SiH2 to give the thorium amido hydrido metallocenes [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiH2Ph] (2) and [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiHPh2] (3), respectively. Complex 2 readily inserts unsaturated molecules into the Th–H bond, whereas complex 3 reversibly activates an intramolecular aromatic C–H bond to yield [η5-1,2,4-(Me3C)3C5H2]2Th[η2-N,C-{N(p-MeC6H3)(SiHPh2)}] (4) and H2. The experimental results have been complemented by density functional theory (DFT) studies and provide a detailed understanding of the observed reactivity. In addition, a comparison between Th and early transition metals reveals that the Th4+ behaves more like an actinide than a transition metal.
N(p-tolyl) (1) activates Si–H bonds in PhSiH3 or Ph2SiH2 to give the thorium amido hydrido metallocenes [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiH2Ph] (2) and [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiHPh2] (3), respectively. Complex 2 readily inserts unsaturated molecules into the Th–H bond, whereas complex 3 reversibly activates an intramolecular aromatic C–H bond to yield [η5-1,2,4-(Me3C)3C5H2]2Th[η2-N,C-{N(p-MeC6H3)(SiHPh2)}] (4) and H2. The experimental results have been complemented by density functional theory (DFT) studies and provide a detailed understanding of the observed reactivity. In addition, a comparison between Th and early transition metals reveals that the Th4+ behaves more like an actinide than a transition metal.
Introduction
Terminal imido complexes of actinide-metals containing an AnN functionality have received widespread attention over the last 20 years because of their unique structural properties and their potential application in group transfer reactions and catalysis.1–3 Whereas many uranium imido complexes have been prepared and structurally characterized, only a few of them show significant reactivity.2h–k,s,t,x,3f,g,j,l,o,y In contrast, thorium imido complexes have remained rare and not much is known about their reaction chemistry.2d,3h,k,q This is surprising since thorium has a 7s2 6d2 ground state electron configuration, and one might expect a similar reactivity to that of group 3 and 4 metals, such as Sc, Ti, Zr and Hf, for which several complexes with MN bond have been prepared.4,5 However, the underlying question remains whether 5f-orbitals contribute to the bonding in thorium organometallics and whether Th4+ should be considered as a transition metal or as an actinide element.6 To answer this question we have recently prepared the base-free terminal thorium imido complex [η5-1,2,4-(Me3C)3C5H2]2ThN(p-tolyl) (1).7,8 Complex 1 shows a rich reaction chemistry such as the activation of elemental sulfur (S8).7 Furthermore it is an important intermediate in the catalytic hydroamination of internal acetylenes,7 an efficient catalyst for the trimerization of PhCN,7 and a useful precursor for the preparation of oxido and sulfido thorium metallocenes [η5-1,2,4-(Me3C)3C5H2]2ThE (E = O, S) by cycloaddition–elimination reactions with Ph2CE (E = O, S) or CS2.9 Encouraged by this broad reactivity, we are now focusing on small molecule activation. Herein, the first Si–H bond activations by an actinide imido complex and the reactivity of the resulting thorium amido hydrido complexes are reported. Furthermore, the difference between thorium and early transition metal imido complexes will be addressed.
Experimental
General methods
All reactions and product manipulations were carried out under an atmosphere of dry dinitrogen with rigid exclusion of air and moisture using standard Schlenk or cannula techniques, or in a glove box. All organic solvents were freshly distilled from sodium benzophenone ketyl immediately prior to use. PhSiH3 and Ph2SiH2 were freshly distilled from CaH2 immediately prior to use. [η5-1,2,4-(Me3C)3C5H2]2ThN(p-tolyl) (1) was prepared according to literature methods.7 All other chemicals were purchased from Aldrich Chemical Co. and Beijing Chemical Co. and used as received unless otherwise noted. Infrared spectra were obtained from KBr pellets on an Avatar 360 Fourier transform spectrometer. 1H and 13C NMR spectra were recorded on a Bruker AV 400 spectrometer at 400 and 100 MHz, respectively. All chemical shifts are reported in δ units with reference to the residual protons of the deuterated solvents, which are internal standards, for proton and carbon chemical shifts. Melting points were measured on an X-6 melting point apparatus and were uncorrected. Elemental analyses were performed on a Vario EL elemental analyzer.
Syntheses
Method A. A toluene (5 mL) solution of PhSiH3 (68 mg, 0.622 mmol) was added to a toluene (10 mL) solution of [η5-1,2,4-(Me3C)3C5H2]2Th
N(p-tolyl) (1; 500 mg, 0.622 mmol). After this mixture was stirred at room temperature for one hour, the solvent was removed. The residue was extracted with n-hexane (10 mL × 2) and filtered. The volume of the filtrate was reduced to ca. 2 mL and colorless crystals of 2a were isolated when this solution stood at room temperature for 2 days. Yield: 465 mg (82%) (found: C, 61.72; H, 8.18; N, 1.63. C47H73NSiTh requires C, 61.88; H, 8.07; N, 1.54%). Mp: 116–118 °C (decomp.). 1H NMR (C6D6): δ 18.07 (s, 1H, ThH), 7.59 (d, J = 5.6 Hz, 2H, aryl), 7.13 (m, 3H, aryl), 7.06 (d, J = 5.6 Hz, 2H, aryl), 7.02 (m, 2H, aryl), 6.14 (d, J = 3.6 Hz, 2H, ring CH), 6.01 (d, J = 3.6 Hz, 2H, ring CH), 5.06 (s, 2H, SiH2), 2.15 (s, 3H, tolylCH3), 1.57 (s, 18H, (CH3)3C), 1.48 (s, 18H, (CH3)3C), 1.43 (s, 18H, (CH3)3C). 13C{1H} NMR (C6D6): δ 143.1 (phenyl C), 142.2 (phenyl C), 135.9 (phenyl C), 135.3 (phenyl C), 134.9 (phenyl C), 130.0 (phenyl C), 129.6 (phenyl C), 122.9 (phenyl C), 119.0 (ring C), 115.7 (ring C), 115.5 (ring C), 115.3 (ring C), 113.7 (ring C), 35.1 ((CH3)3C), 34.6 ((CH3)3C), 34.1 ((CH3)3C), 34.0 ((CH3)3C), 33.7 ((CH3)3C), 33.2 ((CH3)3C), 20.4 (CH3). IR (KBr, cm−1): ν 2959 (s, C–H), 2124 (w, Si–H), 1606 (m, CC), 1458 (s, C–C), 1385 (s, C–C), 1258 (s, C–N), 1094 (s, C–N), 1019 (s, C–N). The NMR sample was maintained at 70 °C and monitored periodically by 1H NMR spectroscopy. After 4 days, resonances of the anti-isomer [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiH2Ph] (2b) (1H NMR (C6D6): δ 16.93 (s, 1H, ThH), 8.00 (s, 1H, aryl), 7.92 (d, J = 6.8 Hz, 2H, aryl), 7.39 (d, J = 7.4 Hz, 2H, aryl), 7.23 (d, J = 7.4 Hz, 2H, aryl), 7.13 (m, 2H, aryl), 6.41 (d, J = 3.2 Hz, 2H, ring CH), 6.20 (d, J = 3.2 Hz, 2H, ring CH), 5.23 (s, 2H, SiH2), 2.38 (s, 3H, tolylCH3), 1.39 (s, 18H, (CH3)3C), 1.38 (s, 18H, (CH3)3C), 1.36 (s, 18H, (CH3)3C)) were observed by 1H NMR spectroscopy (syn/anti ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9). However, on prolonged heating some degradation was also observed.
:9). However, on prolonged heating some degradation was also observed.
Method B.
NMR scale. To a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th
N(p-tolyl) (1; 16 mg, 0.02 mmol) and C6D6 (0.5 mL), an excess of PhSiH3 was added. Upon addition complete conversion to 2a was observed by 1H NMR spectroscopy.
Method A. This compound was prepared as colorless microcrystals from the reaction of [η5-1,2,4-(Me3C)3C5H2]2Th
N(p-tolyl) (1; 500 mg, 0.622 mmol) and Ph2SiH2 (114 mg, 0.622 mmol) in toluene (15 mL) and recrystallization from an n-hexane solution by a similar procedure as outlined for the synthesis of 2a. Yield: 516 mg (84%) (found: C, 64.52; H, 8.02; N, 1.43. C53H77NSiTh requires C, 64.41; H, 7.85; N, 1.42%). Mp: 120–122 °C (decomp.). 1H NMR (C6D6): δ 17.84 (s, 1H, ThH), 7.74 (s, 2H, aryl), 7.50 (d, J = 7.2 Hz, 2H, aryl), 7.11 (m, 8H, aryl), 7.03 (d, J = 7.2 Hz, 2H, aryl), 6.02 (s, 4H, ring CH), 5.08 (s, 1H, SiH), 2.18 (s, 3H, tolylCH3), 1.58 (s, 18H, (CH3)3C), 1.43 (s, 36H, (CH3)3C). 13C{1H} NMR (C6D6): δ 136.2 (phenyl C), 135.8 (phenyl C), 134.5 (phenyl C), 131.5 (phenyl C), 129.9 (phenyl C), 129.7 (phenyl C), 129.6 (phenyl C), 128.2 (phenyl C), 127.8 (ring C), 127.5 (ring C), 119.0 (ring C), 115.5 (ring C), 115.2 (ring C), 35.0 ((CH3)3C), 34.8 ((CH3)3C), 34.4 ((CH3)3C), 34.1 ((CH3)3C), 33.3 ((CH3)3C), 33.1 ((CH3)3C), 20.5 (CH3). IR (KBr, cm−1): ν 2961 (s, C–H), 2110 (w, Si–H), 1587 (m, CC), 1454 (m, C–C), 1384 (s, C–C), 1260 (s, C–N), 1090 (s, C–N), 1018 (s, C–N). The NMR sample was maintained at 70 °C and monitored periodically by 1H NMR spectroscopy. After 1 h, new resonances of [η5-1,2,4-(Me3C)3C5H2]2Th[η2-N,C-{N(p-MeC6H3)(SiHPh2)}] (4) (see below) were observed by 1H NMR spectroscopy with 20% conversion. After 3 h, 27% conversion was observed. However, no change in this ratio was detected on prolonged heating.
Method B.
NMR scale. To a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th
N(p-tolyl) (1; 16 mg, 0.02 mmol) and C6D6 (0.5 mL), an excess of Ph2SiH2 was added. Complete conversion to 3a was observed by 1H NMR spectroscopy.
NMR scale. To a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th[η2-N,C-{N(p-MeC6H3)(SiHPh2)}] (4; 20 mg, 0.02 mmol) and C6D6 (0.5 mL) an excess of H2 (at 1 atm) was added. 1H NMR spectroscopy indicated complete conversion to 3a and 3b (ratio 3a/3bca. 10
:1; 1H NMR (C6D6): δ 16.84 (s, 1H, ThH), 7.84 (d, J = 6.8 Hz, 1H, aryl), 7.60 (d, J = 7.5 Hz, 2H, aryl), 7.34 (s, 2H, aryl), 7.11 (m, 6H, aryl), 6.95 (s, 1H, aryl H), 6.77 (d, J = 8.1 Hz, 2H, aryl), 6.21 (d, J = 3.0 Hz, 2H, ring CH), 6.18 (d, J = 3.0 Hz, 2H, ring CH), 5.14 (s, 1H, SiH), 2.19 (s, 3H, tolylCH3), 1.54 (s, 18H, (CH3)3C), 1.41 (s, 18H, (CH3)3C), 1.39 (s, 18H, (CH3)3C)).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) NC6H11)–N(C6H11)] (6).
NC6H11)–N(C6H11)] (6).
Method A. A toluene (5 mL) solution of N,N′-dicyclohexylcarbodiimide (DCC; 103 mg, 0.50 mmol) was added to a toluene (10 mL) solution of [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiH2Ph] (2a; 228 mg, 0.25 mmol). After this solution was stirred at room temperature for 2 hours, the solvent was removed. The residue was extracted with n-hexane (10 mL × 2) and filtered. The volume of the filtrate was reduced to ca. 2 mL, and colorless crystals of 6 were isolated when this solution stood at room temperature for 2 days. Yield 217 mg (86%) (found: C, 64.32; H, 8.58; N, 4.03. C54H87N3Th requires C, 64.19; H, 8.68; N, 4.16%). Mp: 210–212 °C (decomp.). 1H NMR (C6D6): δ 7.30 (d, J = 8.0 Hz, 2H, aryl), 7.09 (d, J = 8.0 Hz, 2H, aryl), 6.68 (s, 2H, ring CH), 6.35 (s, 2H, ring CH), 3.84 (m, 1H, NCH), 3.31 (m, 2H, CH2), 3.24 (m, 1H, NCH), 2.36 (m, 2H, CH2), 2.25 (s, 3H, tolylCH3), 2.12 (m, 4H, CH2), 1.84 (m, 8H, CH2), 1.53 (m, 4H, CH2), 1.52 (s, 18H, (CH3)3C), 1.50 (s, 18H, (CH3)3C), 1.27 (s, 18H, (CH3)3C). 13C{1H} NMR (C6D6): δ 152.0 (C
N), 145.9 (phenyl C), 144.2 (phenyl C), 143.6 (phenyl C), 143.6 (phenyl C), 130.8 (ring C), 129.3 (ring C), 127.5 (ring C), 125.9 (ring C), 118.3 (ring C), 62.1 (CHN), 53.8 (CHN), 36.4 ((CH3)3C), 35.0 ((CH3)3C), 34.9 ((CH3)3C), 34.8 ((CH3)3C), 34.5 ((CH3)3C), 33.2 ((CH3)3C), 32.4 (CH2), 31.2 (CH2), 28.0 (CH2), 27.2 (CH2), 27.1 (CH2), 25.6 (CH2), 20.8 (CH3). IR (KBr, cm−1): ν 2961 (s, C–H), 1618 (m, CN), 1574 (s, CC), 1448 (s, C–C), 1385 (s, C–C), 1259 (s, C–N), 1093 (s, C–N), 1019 (s, C–N).
Method B.
NMR scale. To a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiH2Ph] (2a; 18 mg, 0.02 mmol) and C6D6 (0.5 mL), DCC (8.2 mg, 0.04 mmol) was added. Complete conversion to 6 and (C6H11)N
CHN(C6H11)(SiH2Ph) (5) (1H NMR (C6D6): δ 7.37 (s, 1H, NCH), 7.10 (m, 5H, aryl), 4.22 (s, 2H, SiH2), 3.12 (m, 2H, NCH), 1.84 (m, 16H, CH2), 1.32 (m, 4H, CH2)) was observed by 1H NMR spectroscopy.
Method C.
NMR scale. To a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th
N(p-tolyl) (1; 16 mg, 0.02 mmol) and C6D6 (0.5 mL), DCC (4.1 mg, 0.02 mmol) was added. Complete conversion to 6 was observed by 1H NMR spectroscopy.
NMR scale. To a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiH2Ph] (2a; 18 mg, 0.02 mmol) and C6D6 (0.5 mL), DCC (4.1 mg, 0.02 mmol) was added. Resonances due to 6 along with those of 5 and unreacted 2a were observed by 1H NMR spectroscopy (50% conversion based on 2a).
X-ray crystallography
Single-crystal X-ray diffraction measurements were carried out on a Bruker SMART CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). An empirical absorption correction was applied using the SADABS program.10 All structures were solved by direct methods and refined by full-matrix least squares on F2 using the SHELXL-97 program package.11 The hydride atom in 2a was located from a difference-Fourier map and refined isotropically. Hydrogen atoms were geometrically fixed using the riding model. Disordered solvents in the voids of 2a and 6 were modeled or removed by using the SQUEEZE program.12 Crystallographic details for 2a, 4 and 6 are summarized in Table 1.| Compound | 2a | 4 | 6 |
|---|---|---|---|
| Formula | C47H73NSiTh | C53H75NSiTh | C54H87N3Th |
| Fw | 912.19 | 986.27 | 1010.31 |
| Crystal system | Orthorhombic | Monoclinic | Triclinic |
| Space group | Pbca | P21/c |
P(![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) ) |
| a (Å) | 10.425 (1) | 16.578 (3) | 10.436 (2) |
| b (Å) | 20.765 (1) | 16.572 (3) | 15.509 (3) |
| c (Å) | 44.289 (3) | 17.515 (3) | 20.314 (4) |
| α (deg.) | 90 | 90 | 92.79 (1) |
| β (deg.) | 90 | 97.936 (3) | 92.25 (1) |
| γ (deg.) | 90 | 90 | 109.07 (1) |
| V (Å3) | 9587.6 (10) | 4765.9 (13) | 3098.4 (10) |
| Z | 8 | 4 | 2 |
| D calc (g cm−3) | 1.264 | 1.375 | 1.083 |
| μ (Mo/Kα)calc (cm−1) | 3.164 | 3.188 | 2.436 |
| Size (mm) | 0.39 × 0.21 × 0.11 | 0.25 × 0.18 × 0.10 | 0.45 × 0.20 × 0.11 |
| F(000) | 3728 | 2016 | 1044 |
| 2θ range (deg.) | 3.92 to 55.34 | 4.40 to 55.22 | 3.54 to 50.50 |
| No. of reflns, collected | 11151 |
26706 |
10908 |
| No. of obsd reflns | 8989 | 7619 | 9578 |
| No. of variables | 528 | 527 | 542 |
| Abscorr (Tmax, Tmin) | 0.72, 0.37 | 0.74, 0.50 | 0.78, 0.41 |
| R | 0.032 | 0.051 | 0.078 |
| R w | 0.064 | 0.121 | 0.224 |
| R all | 0.066 | 0.146 | 0.230 |
| Gof | 1.10 | 1.07 | 1.12 |
| CCDC | 977309 | 977310 | 977311 |
Computational methods
All calculations were carried out with the Gaussian 09 program (G09),13 employing the B3PW91 method, plus polarizable continuum model (PCM) and D3 (ref. 14) (denoted as B3PW91-PCM + D3), with standard 6-31G(d) basis set for C, H, N and Si elements and Stuttgart RLC ECP from EMSL basis set exchange (https://bse.pnl.gov/bse/portal) for Th element,15 to fully optimize the geometries of reactants, complexes, transition state, intermediates, and product structures, and to mimic experimental toluene-solvent conditions (dielectricity constant ε = 2.379). All stationary points were subsequently characterized by vibrational analyses, from which their respective zero-point (vibrational) energy (ZPE) were extracted and used in the relative energy determinations; in addition to ensure that the reactant, complex, intermediate, product and transition state structures resided at minima and 1st order saddle points, respectively, on their potential energy hyper surfaces.Results and discussion
Complex 1 reacts rapidly with silanes such as PhSiH3 or Ph2SiH2 to give the amido hydrido metallocenes [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiH2Ph] (2a) and [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiHPh2] (3a), respectively (Scheme 1). Consistent with a Si–H bond addition across the ThN bond the syn-isomer is formed exclusively. However, in contrast to scandium4f and titanium5k,l imido complexes, the reaction of 1 with silanes is irreversible, which is presumably a consequence of the more polarized actinide imido bond (see ESI†). Previous studies have clearly established that the 5f orbitals play a key role in the bonding of actinide complexes, which results in a very polarized AnE bond, whereas this is not the case for group 4 metal complexes.6c Therefore, actinide AnE bonds exhibit different reactivity patterns compared to those of early transition metals, as illustrated by reactivity of imido complexes (MNR) with silanes, in which the less polarized MN bond (M is early transition metal) shows a reversible Si–H addition, whereas the more polarized ThN bond undergoes an irreversible Si–H addition because of a more pronounced thermodynamic stabilization of the Si–H addition products. Heating of 2a at 70 °C for four days forms the anti-isomer 2b (ca. 10%). Under similar reaction conditions, complex 3a is in equilibrium with the aromatic C–H bond activation product [η5-1,2,4-(Me3C)3C5H2]2Th[η2-N,C-{N(p-MeC6H3)(SiHPh2)}] (4) and H2 (Scheme 1). Moreover, the addition of H2 (1 atm) to 4 at room temperature results in a mixture of the syn-/anti-isomers 3a and 3b in a 10:1 ratio (Scheme 1), as established by 1H NMR spectroscopy. Although complex 1 reacts rapidly with PhSiH3 or Ph2SiH2, no reaction was observed with tertiary silanes such as Et3SiH and Ph3SiH most likely because of the steric hindrance.
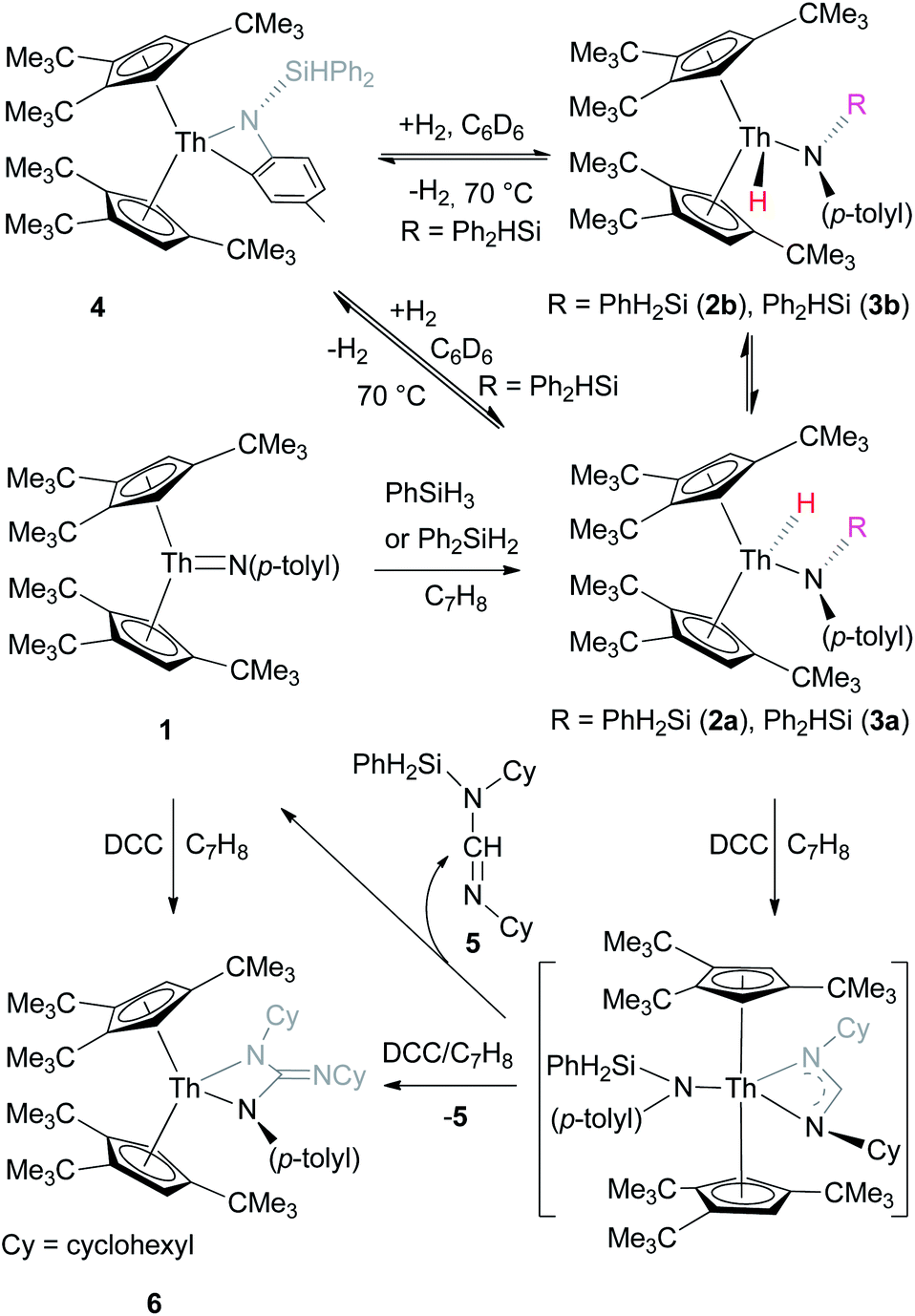 | ||
| Scheme 1 Synthesis of compounds 2–4 and 6. | ||
The molecular structure of [η5-1,2,4-(Me3C)3C5H2]2Th(H)[N(p-tolyl)SiH2Ph] (2a) is shown in Fig. 1. The cyclopentadienyl rings adopt a nearly eclipsed conformation with an average Th–C (ring) distance of 2.858(3) Å, and a Cp (cent)–Th–Cp (cent) angle of 138.1(3)°. The Th–N distance of 2.387(2) Å is comparable to the values found in [η5-1,2,4-(Me3C)3C5H2]2Th[(bipy)(SCPh2)] (2.435(1) Å)16 and [η5-1,2,4-(Me3C)3C5H2]2Th(N-p-tolyl)2 (2.228(3) Å).7 The Th–H distance is 2.01(3) Å, which is in line with reported terminal and bridging Th–H distances, (2,6-tBu2C6H3O)6Th3(μ3-H)2(μ-H)4 (2.0(1)–2.6(1) Å),17 [(η5-C5Me5)2ThH(μ-H)]2 (2.03(1) (terminal)–2.29(3) (bridging) Å),18 (η5-C5Me5)3ThH (2.33(13) Å),19 [η5-1,3-(Me3C)2C5H3]3ThH (1.99(5) Å),20 and (η5-C5Me4H)4[μ-η5-C5Me3H(CH2)-κC]2Th4(μ-H)4(μ3-H)4 (2.503(4) and 2.508(4) Å).21
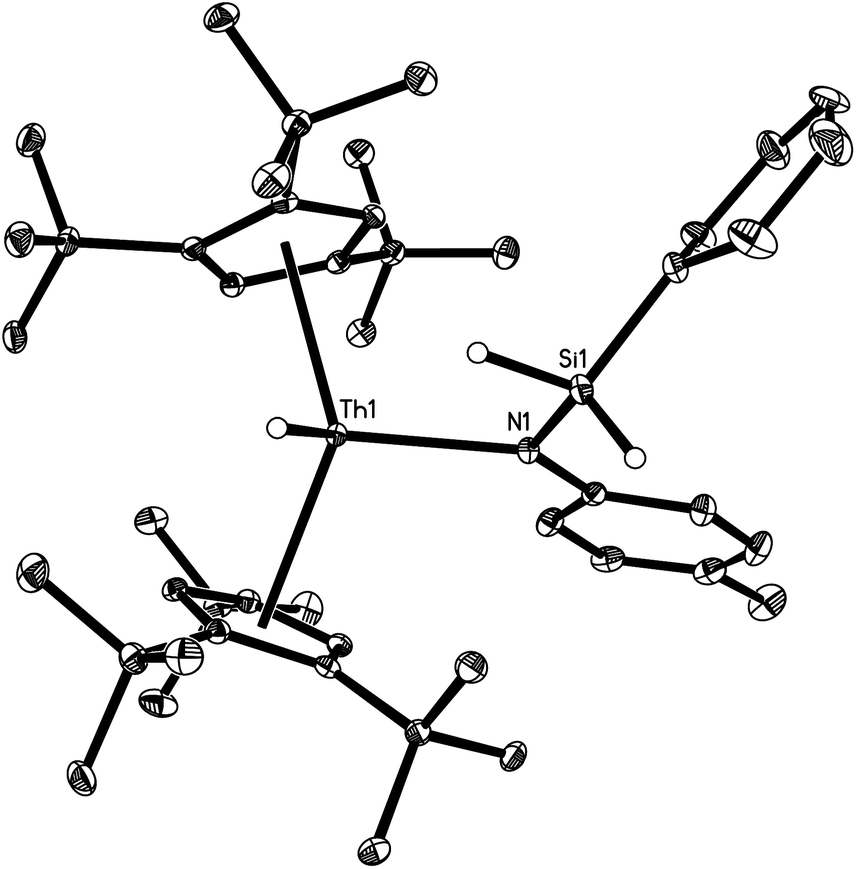 | ||
| Fig. 1 Molecular structure of 2a (thermal ellipsoids drawn at the 35% probability level). Selected bond lengths [Å] and angles [°]: Th–C(Cp) (av.) 2.858(3), Th–C(Cp) (range) 2.870(3) to 2.946(3), Th–Cp(cent) (av.) 2.592(3), Th–N 2.387(2), Th–H 2.01(3), Th–Si 3.256(1), Cp (cent)–Th–Cp (cent) 138.1(3), N–Th–H 111.9(11). | ||
Fig. 2 shows the molecular structure of [η5-1,2,4-(Me3C)3C5H2]2Th[η2-N,C-{N(p-MeC6H3)(SiHPh2)}] (4). The average Th–C (ring) distance is 2.882(6) Å, and the Cp (cent)–Th–Cp (cent) angle is 140.6(2)°. The Th–N distance of 2.310(5) Å is shorter than that (2.387(2) Å found in 2a. Furthermore, the Th–C(40) distance of 2.420(3) Å is also significantly shorter than the reported Th–Caryl bond distances (2.548(2)–2.654(14) Å).22–24 Interestingly, only four structurally characterized thorium aryl complexes have been reported, and in all of them the thorium–aryl bond is supported by chelating ligands.22–24
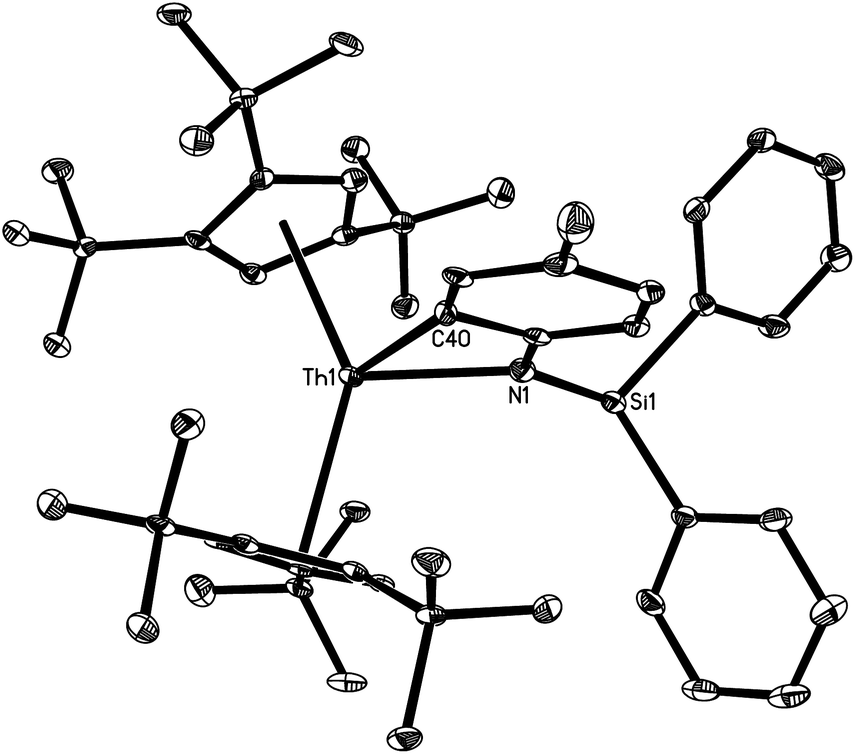 | ||
| Fig. 2 Molecular structure of 4 (thermal ellipsoids drawn at the 35% probability level). Selected bond lengths [Å] and angles [°]: Th–C(Cp) (av.) 2.882(6), Th–C(Cp) (range) 2.803(6) to 2.956(6), Th–Cp (cent) (av.) 2.617(6), Th–N(1) 2.310(5), Th–C(40) 2.420(7), Cp (cent)–Th–Cp (cent) 140.6(2), N(1)–Th–C(40) 61.8(2). | ||
As demonstrated above, complex 1 irreversibly activates Si–H bonds to yield the amido hydrido complexes 2 and 3. Complex 3 undergoes a reversible intramolecular C–H bond activation to give 4 and H2. DFT calculations were performed at the B3PW91 level of theory to further understand the observed process. To properly describe the observed reactivity it was necessary to include solvent and dispersion effects (see ESI†). The reaction of 1 with Ph2SiH2 to 3a is exergonic with ΔG (343 K) = −17.6 kcal mol−1 and proceeds concerted via the 4-membered transition state TS formed by the ThN and Si–H moieties. The Si–N and Th–H bonds in TS are 2.343 and 2.299 Å, respectively, and ca. 0.61 and 0.23 Å longer than those in the product 3a (see ESI†). The barrier for this reaction is ΔG‡ = 16.7 kcal mol−1 (343 K) (15.2 kcal mol−1 at 298 K) (Fig. 3); and these values are consistent with the rapid and irreversible formation of 3a at ambient temperature. The transformation of 3a to 4 + H2 is more complicated and involves the anti-intermediate 3b and two transition states (TSa and TSb). The barrier for the conversion of 3a to 3bviaTSa is 33.7 kcal mol−1 at 343 K and also represents the rate-limiting step in this reaction. The computed barrier is consistent with the experimental observation that this reaction only occurs at elevated temperatures, and it is also in reasonable agreement with the experimentally estimated barrier of 26.8 kcal mol−1 (see ESI†). The deviation between the computed and experimental values is mainly attributed to the low solubility of H2 in organic solvent, which complicates the kinetic evaluation because the actual H2 concentration cannot be determined with certainty (see ESI†). However, for TSa a closer inspection reveals an interesting feature, since the syn-/anti-intermediates are interconverted not by rotation of the –N(R)(p-tolyl) moiety along the Th–N axis, but by a Th–H movement (Fig. 3). This is a direct consequence of the severe steric bulk of the two 1,2,4-(Me3C)3C5H2 ligands, which hinders the rotation along the Th–N axis. Nevertheless, once complex 3b is formed, H2 is released viaTSb and with a low barrier of 12.6 kcal mol−1 (Fig. 3). This makes the anti-isomer 3b difficult to isolate, since H2 release from the reaction mixture shifts the equilibrium to the final product 4. On the contrary, H2 addition to complex 4 forms a mixture of 3b and 3a.
 | ||
| Fig. 3 Energy and free energy profile (kcal mol−1) for the reaction of 1 with Ph2SiH2, obtained with B3PW91-PCM + D3 method. [Th] = [η5-1,2,4-(Me3C)3C5H2]2Th. R = p-tolyl. | ||
Furthermore, similar to the amido hydrido scandium complex,4f amido hydrido thorium complexes can also insert unsaturated substrates such as carbodiimides into the Th–H bond. For example, treatment of 2a with 2 equiv. of N,N′-dicyclohexylcarbodiimide (DCC) rapidly forms the metallacycle [η5-1,2,4-(Me3C)3C5H2]2Th[N(p-tolyl)C(NC6H11)–N(C6H11)] (6) and azomethine (C6H11)NCHN(C6H11)(SiH2Ph) (5) in quantitative conversion (Scheme 1). Furthermore, analogous to group 4 imido complexes,25 thorium imido 1 can react with DCC to yield metallacycle 6 (Scheme 1), whereas 2a reacts with 1 equiv. of DCC to give 6 in only 50% conversion. These observations suggest that the insertion product [η5-1,2,4-(Me3C)3C5H2]2Th[N(p-tolyl)SiHPh2][η3-N(C6H11)CHN(C6H11)] is unstable and eliminates azomethine 5 to form the imido complex 1, which then reacts with DCC to the metallacycle 6 by an [2 + 2] cycloaddition reaction (Scheme 1).
The molecular structure of [η5-1,2,4-(Me3C)3C5H2]2Th[N(p-tolyl)C(NC6H11)–N(C6H11)] (6) is shown in Fig. 4. The Th4+ ion is η5-bound to two Cp-rings and σ-coordinate to the two nitrogen atoms of the [N(p-tolyl)C(NC6H11)–N(C6H11)] group in a distorted-tetrahedral geometry with an averaged Th–C (ring) distance of 2.877(11) Å. The dihedral angle defined by the planes containing the Cp (cent)–Th–Cp (cent) and N(1)–C(42)–N(3) is 75.2(3)°, and the N(1)–Th(1)–N(3) angle is 58.5(3)°. These metric parameters resemble those found in the alkyne addition complexes [η5-1,2,4-(Me3C)3C5H2]2Th[N(p-tolyl)C(R′)C(R′)] (R′ = Me, Ph),7 while the Th–N distances (2.346(9) and 2.337(8) Å) are similar to those found in 2a and 4.
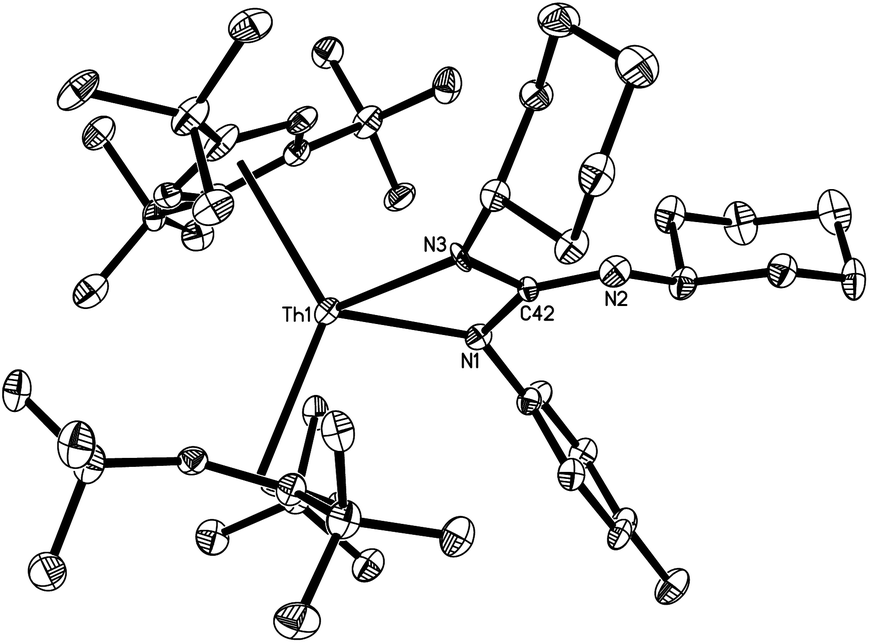 | ||
| Fig. 4 Molecular structure of 6 (thermal ellipsoids drawn at the 35% probability level). Selected bond lengths [Å] and angles [°]: Th–C(Cp) (av.) 2.877(11), Th–C(Cp) (range) 2.772(10) to 3.024(11), Th–Cp (cent) (av.) 2.613(9), Th–N(1) 2.346(9), Th–N(3) 2.337(8), Cp (cent)–Th–Cp (cent) 128.6(3), N(1)–Th–N(3) 58.5(3). | ||
Conclusions
In conclusion, the first example of a Si–H bond activation by a terminal actinide imido complex has been comprehensively studied. In contrast to scandium4f and titanium5k,l imido complexes, silanes such as PhSiH3 and Ph2SiH2 add irreversibly to the thorium imido [η5-1,2,4-(Me3C)3C5H2]2ThN(p-tolyl) (1), supporting the notion that Th4+ behaves more like an actinide than a transition metal.6c DFT studies reveal that the 1,2-addition proceeds in a concerted, 4-membered transition state to give amido hydrido complexes 2 and 3. These compounds are reactive species, as illustrated by the formation of H2 and [η5-1,2,4-(Me3C)3C5H2]2Th[η2-N,C-{N(p-MeC6H3)(SiHPh2)}] (4) via an intramolecular aromatic C–H bond activation in 3, and by the insertion of unsaturated substrates such as DCC into the Th–H bond of 2. The development of new actinide imido complexes and the exploration of thorium amido hydrido complexes in catalysis are ongoing projects in these laboratories.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21172022, 21302155, 21373030), and the Deutsche Forschungsgemeinschaft (DFG) through the Emmy-Noether program (WA 2513/2). We thank Dr Xuebin Deng for his help with the crystallography, and Prof. Richard A. Andersen for helpful discussions.Notes and references
- For selected recent reviews, see: (a) M. Ephritikhine, Dalton Trans., 2006, 2501–2516 RSC; (b) E. Barnea and M. S. Eisen, Coord. Chem. Rev., 2006, 250, 855–899 CrossRef CAS PubMed; (c) A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341–349 CrossRef CAS PubMed; (d) T. Andrea and M. S. Eisen, Chem. Soc. Rev., 2008, 37, 550–567 RSC; (e) C. R. Graves and J. L. Kiplinger, Chem. Commun., 2009, 3831–3853 RSC; (f) T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC; (g) P. L. Arnold, Chem. Commun., 2011, 47, 9005–9010 RSC; (h) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (i) T. W. Hayton, Nat. Chem., 2013, 5, 451–452 CrossRef CAS PubMed.
- For selected papers on actinide non-metallocenes containing terminal imido groups, see: (a) C. J. Burns, W. H. Smith, J. C. Huffman and A. P. Sattelberger, J. Am. Chem. Soc., 1990, 112, 3237–3239 CrossRef CAS; (b) D. R. Brown and R. G. Denning, Inorg. Chem., 1996, 35, 6158–6163 CrossRef CAS; (c) P. Roussel, R. Boaretto, A. J. Kingsley, N. W. Alcock and P. Scott, J. Chem. Soc., Dalton Trans., 2002, 1423–1428 RSC; (d) I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2003, 42, 814–818 CrossRef CAS PubMed; (e) I. Castro-Rodriguez, K. Olsen, P. Gantzel and K. Meyer, J. Am. Chem. Soc., 2003, 125, 4565–4571 CrossRef CAS PubMed; (f) T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941–1943 CrossRef CAS PubMed; (g) T. W. Hayton, J. M. Boncella, B. L. Scott, E. R. Batista and P. J. Hay, J. Am. Chem. Soc., 2006, 128, 10549–10559 CrossRef CAS PubMed; (h) T. W. Hayton, J. M. Boncella, B. L. Scott and E. R. Batista, J. Am. Chem. Soc., 2006, 128, 12622–12623 CrossRef CAS PubMed; (i) I. Castro-Rodriguez, H. Nakai and K. Meyer, Angew. Chem., Int. Ed., 2006, 45, 2389–2392 CrossRef CAS PubMed; (j) L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, J. Am. Chem. Soc., 2008, 130, 2930–2931 CrossRef CAS PubMed; (k) S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed; (l) L. P. Spencer, E. J. Schelter, P. Yang, R. L. Gdula, B. L. Scott, J. D. Thompson, J. L. Kiplinger, E. R. Batista and J. M. Boncella, Angew. Chem., Int. Ed., 2009, 48, 3795–3798 CrossRef CAS PubMed; (m) L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, Inorg. Chem., 2009, 48, 2693–2700 CrossRef CAS PubMed; (n) L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, Inorg. Chem., 2009, 48, 11615–11623 CrossRef CAS PubMed; (o) T. Cantat, C. R. Graves, B. L. Scott and J. L. Kiplinger, Angew. Chem., Int. Ed., 2009, 48, 3681–3684 CrossRef CAS PubMed; (p) D. L. Swartz II, L. P. Spencer, B. L. Scott, A. L. Odom and J. M. Boncella, Dalton Trans., 2010, 39, 6841–6846 RSC; (q) L. A. Seaman, S. Fortier, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 636–646 CrossRef CAS PubMed; (r) R. E. Jilek, L. P. Spencer, D. L. Kuiper, B. L. Scott, U. J. Williams, J. M. Kikkawa, E. J. Schelter and J. M. Boncella, Inorg. Chem., 2011, 50, 4235–4237 CrossRef CAS PubMed; (s) E. M. Matson, P. E. Fanwick and S. C. Bart, Eur. J. Inorg. Chem., 2012, 5471–5478 CrossRef CAS; (t) E. M. Matson, M. G. Crestani, P. E. Fanwick and S. C. Bart, Dalton Trans., 2012, 41, 7952–7958 RSC; (u) O. P. Lam, S. M. Franke, H. Nakai, F. W. Heinemann, W. Hieringer and K. Meyer, Inorg. Chem., 2012, 51, 6190–6199 CrossRef CAS PubMed; (v) C. Camp, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed; (w) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed; (x) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed.
- For selected papers about actinide metallocenes containing terminal imido groups, see: (a) R. E. Cramer, K. Panchanatheswaran and J. W. Gilje, J. Am. Chem. Soc., 1984, 106, 1853–1854 CrossRef CAS; (b) J. G. Brennan and R. A. Andersen, J. Am. Chem. Soc., 1985, 107, 514–516 CrossRef CAS; (c) R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS; (d) D. S. J. Arney, C. J. Burns and D. C. Smith, J. Am. Chem. Soc., 1992, 114, 10068–10069 CrossRef CAS; (e) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1993, 115, 9840–9841 CrossRef CAS; (f) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS; (g) T. Straub, W. Frank, G. J. Reiss and M. S. Eisen, J. Chem. Soc., Dalton Trans., 1996, 2541–2546 RSC; (h) A. Haskel, T. Straub and M. S. Eisen, Organometallics, 1996, 15, 3773–3775 CrossRef CAS; (i) B. P. Warner, B. L. Scott and C. J. Burns, Angew. Chem., Int. Ed., 1998, 37, 959–960 CrossRef CAS; (j) R. G. Peters, B. P. Warner, B. L. Scott and C. J. Burns, Organometallics, 1999, 18, 2587–2589 CrossRef CAS; (k) T. Straub, A. Haskel, T. G. Neyroud, M. Kapon, M. Botoshansky and M. S. Eisen, Organometallics, 2001, 20, 5017–5035 CrossRef CAS; (l) J. L. Kiplinger, D. E. Morris, B. L. Scott and C. J. Burns, Chem. Commun., 2002, 30–31 RSC; (m) W. J. Evans, S. A. Kozimor and J. W. Ziller, Chem. Commun., 2005, 4681–4683 RSC; (n) G. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4262 CrossRef CAS; (o) G. Zi, L. L. Blosch, L. Jia and R. A. Andersen, Organometallics, 2005, 24, 4602–4612 CrossRef CAS; (p) C. R. Graves, B. L. Scott, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2007, 129, 11914–11915 CrossRef CAS PubMed; (q) E. J. Schelter, D. E. Morris, B. L. Scott and J. L. Kiplinger, Chem. Commun., 2007, 1029–1031 RSC; (r) C. R. Graves, P. Yang, S. A. Kozimor, A. E. Vaughn, D. L. Clark, S. D. Conradson, E. J. Schelter, B. L. Scott, J. D. Thompson, P. J. Hay, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 5272–5285 CrossRef CAS PubMed; (s) C. R. Graves, B. L. Scott, D. E. Morris and J. L. Kiplinger, Organometallics, 2008, 27, 3335–3337 CrossRef CAS; (t) C. R. Graves, A. E. Vaughn, E. J. Schelter, B. L. Scott, J. D. Thompson, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2008, 47, 11879–11891 CrossRef CAS PubMed; (u) L. P. Spencer, R. L. Gdula, T. W. Hayton, B. L. Scott and J. M. Boncella, Chem. Commun., 2008, 4986–4988 RSC; (v) W. J. Evans, C. A. Traina and J. W. Ziller, J. Am. Chem. Soc., 2009, 131, 17473–17481 CrossRef CAS PubMed; (w) C. R. Graves, B. L. Scott, D. E. Morris and J. L. Kiplinger, Chem. Commun., 2009, 776–778 RSC; (x) W. J. Evans, E. Montalvo, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2010, 49, 222–228 CrossRef CAS PubMed; (y) R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS PubMed.
- Selected papers on scandium imido complexes: (a) E. Lu, Y. Li and Y. Chen, Chem. Commun., 2010, 46, 4469–4471 RSC; (b) E. Lu, J. Chu, M. Borzov, Y. Chen and G. Li, Chem. Commun., 2011, 47, 743–745 RSC; (c) J. Chu, E. Lu, Z. Liu, Y. Chen, X. Leng and H. Song, Angew. Chem., Int. Ed., 2011, 50, 7677–7680 CrossRef CAS PubMed; (d) E. Lu, Q. Zhou, Y. Li, J. Chu, Y. Chen, X. Leng and J. Sun, Chem. Commun., 2012, 48, 3403–3405 RSC; (e) Z. Jian, W. Rong, Z. Mou, Y. Pan, H. Xie and D. Cui, Chem. Commun., 2012, 48, 7516–7518 RSC; (f) T. Chu, W. E. Piers, J. L. Dutton and M. Parvez, Organometallics, 2013, 32, 1159–1165 CrossRef CAS; (g) J. Chu, E. Lu, Y. Chen and X. Leng, Organometallics, 2013, 32, 1137–1140 CrossRef CAS; (h) W. Rong, J. Cheng, Z. Mou, H. Xie and D. Cui, Organometallics, 2013, 32, 5523–5529 CrossRef CAS; (i) J. Chu, C. E. Kefalidis, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2013, 135, 8165–8168 CrossRef CAS PubMed.
- Selected papers on group 4 imido complexes: (a) P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 8729–8731 CrossRef CAS; (b) C. C. Cummins, S. M. Baxter and P. T. Wolczanski, J. Am. Chem. Soc., 1988, 110, 8731–8733 CrossRef CAS; (c) J. E. Hill, R. D. Profilet, P. E. Fanwick and I. P. Rothwell, Angew. Chem., Int. Ed., 1990, 29, 664–665 CrossRef; (d) H. W. Roesky, H. Voelker, M. Witt and M. Noltemeyer, Angew. Chem., Int. Ed., 1990, 29, 669–670 CrossRef; (e) P. J. Walsh, F. J. Hollander and R. G. Bergman, Organometallics, 1993, 12, 3705–3723 CrossRef CAS; (f) S. C. Dunn, A. S. Batsanov and P. Mountford, J. Chem. Soc., Chem. Commun., 1994, 2007–2008 RSC; (g) J. L. Bennett and P. T. Wolczanski, J. Am. Chem. Soc., 1994, 116, 2179–2180 CrossRef CAS; (h) J. L. Polse, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 13405–13414 CrossRef CAS; (i) H. M. Hoyt, F. E. Michael and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 1018–1019 CrossRef CAS PubMed; (j) B. Lian, T. P. Spaniol, P. Horrillo-Martínez, K. C. Hultzsch and J. Okuda, Eur. J. Inorg. Chem., 2009, 429–434 CrossRef CAS; (k) P. J. Tiong, A. Nova, E. Clot and P. Mountford, Chem. Commun., 2011, 47, 3147–3149 RSC; (l) P. J. Tiong, A. Nova, A. D. Schwarz, J. D. Selby, E. Clot and P. Mountford, Dalton Trans., 2012, 41, 2277–2288 RSC; (m) A. D. Schwarz, A. J. Nielson, N. Kaltsoyannis and P. Mountford, Chem. Sci., 2012, 3, 819–824 RSC.
- Selected recent papers about the bonding of organoactinide complexes, see: (a) T. Cantat, C. R. Graves, K. C. Jantunen, C. J. Burns, B. L. Scott, E. J. Schelter, D. E. Morris, P. J. Hay and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 17537–17551 CrossRef CAS PubMed; (b) N. Barros, D. Maynau, L. Maron, O. Eisenstein, G. Zi and R. A. Andersen, Organometallics, 2007, 26, 5059–5065 CrossRef CAS; (c) A. Yahia and L. Maron, Organometallics, 2009, 28, 672–679 CrossRef CAS; (d) W. Ren, X. Deng, G. Zi and D.-C. Fang, Dalton Trans., 2011, 40, 9662–9664 RSC.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, Chem.–Eur. J., 2011, 17, 12669–12682 CrossRef CAS PubMed.
- W. Ren, G. Zi and M. D. Walter, Organometallics, 2012, 31, 672–679 CrossRef CAS.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- (a) G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structure from Diffraction Data, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- SQUEEZE: P. V. D. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, Mol. Phys., 1991, 74, 1245–1263 CrossRef.
- W. Ren, W. W. Lukens, G. Zi, L. Maron and M. D. Walter, Chem. Sci., 2013, 4, 1168–1174 RSC.
- D. L. Clark, S. K. Grumbine, B. L. Scott and J. G. Watkin, Organometallics, 1996, 15, 949–957 CrossRef CAS.
- R. W. Broach, A. J. Schultz, J. M. Williams, G. M. Brown, J. M. Manriquez, P. J. Fagan and T. J. Marks, Science, 1979, 203, 172–174 CAS.
- W. J. Evans, G. W. Nyce and J. W. Ziller, Organometallics, 2001, 20, 5489–5491 CrossRef CAS.
- W. Ren, N. Zhao, L. Chen, H. Song and G. Zi, Inorg. Chem. Commun., 2011, 14, 1838–1841 CrossRef CAS PubMed.
- N. A. Siladke, C. L. Webster, J. R. Walensky, M. K. Takase, J. W. Ziller, D. J. Grant, L. Gagliardi and W. J. Evans, Organometallics, 2013, 32, 6522–6531 CrossRef CAS.
- L. A. Seaman, E. A. Pedrick, T. Tsuchiya, G. Wu, E. Jakubikova and T. W. Hayton, Angew. Chem., Int. Ed., 2013, 52, 10589–10592 CrossRef CAS PubMed.
- I. Korobkov, B. Vidjayacoumar, S. I. Gorelsky, P. Billone and S. Gambarotta, Organometallics, 2010, 29, 692–702 CrossRef CAS.
- I. Korobkov, A. Arunachalampillai and S. Gambarotta, Organometallics, 2004, 23, 6248–6252 CrossRef CAS.
- Selected papers on the reaction of group 4 imido complexes with organic imines, see: (a) K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 2669–2670 CrossRef CAS; (b) K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 974–985 CrossRef CAS; (c) R. L. Zuckerman and R. G. Bergman, Organometallics, 2000, 19, 4795–4809 CrossRef CAS; (d) A. E. Guiducci, C. L. Boyd and P. Mountford, Organometallics, 2006, 25, 1167–1187 CrossRef CAS; (e) P. D. Schweizer, H. Wadepohl and L. H. Gade, Organometallics, 2013, 32, 3697–3709 CrossRef CAS; (f) J. Unruangsri, H. Morgan, A. D. Schwarz, A. D. Schofield and P. Mountford, Organometallics, 2013, 32, 3091–3107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NBO analysis of complex 1, cartesian coordinates of all stationary points optimized at B3PW91-PCM + D3 level and kinetic study. CCDC 977309–977311. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc00576g |
| This journal is © The Royal Society of Chemistry 2014 |