 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light alkane oxidation using catalysts prepared by chemical vapour impregnation: tuning alcohol selectivity through catalyst pre-treatment†
Michael M.
Forde
*a,
Robert D.
Armstrong
a,
Rebecca
McVicker
a,
Peter P.
Wells
b,
Nikolaos
Dimitratos
a,
Qian
He
c,
Li
Lu
c,
Robert L.
Jenkins
a,
Ceri
Hammond
a,
Jose Antonio
Lopez-Sanchez‡
a,
Christopher J.
Kiely
c and
Graham J.
Hutchings
a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building Park Place, Cardiff, CF103AT, UK. E-mail: michael.forde@sta.uwi.edu; Fax: +44 (0)2090874030; Tel: +44 (0)2920874059
bUK Catalysis Hub, Research Complex at Harwell, R92 Rutherford Appleton Laboratory, Harwell Science & Innovation Campus, Didcot, OX11 0FA, UK
cDepartment of Materials Science and Engineering, Lehigh University, 5 East Packer Avenue, Bethlehem, PA 18015-3195, USA
First published on 16th June 2014
Abstract
Fe/ZSM-5(30) catalysts have been prepared by chemical vapour impregnation (CVI) using iron(III) acetylacetonate as the precursor. These materials have been used for the oxidation of methane and ethane using aqueous hydrogen peroxide as oxidant. Heating in air leads to materials that exhibit high catalytic activity and give formic and acetic acid with high selectivity from methane and ethane respectively. Heat treatment of the uncalcined materials under a reducing atmosphere results in partial reduction of iron from the FeIII to FeII oxidation state with the majority of the iron being present as isolated octahedral extra-framework species having oxygen neighbours and showing no evidence of a coordination shell containing Al or Fe, as evidenced from studies using X-ray absorption and UV-Vis spectroscopies. These hydrogen treated catalysts show the same catalytic activity as their analogues formed by heating in air, but in contrast exhibit higher alcohol selectivities for both methane and ethane conversion to oxygenates and are reusable. Our findings for both the oxidation of methane and ethane indicate that the selectivity to the oxidation products, i.e. acids or alcohols, can be controlled by tuning the active site structure and/or oxidation state of the Fe species in Fe/ZSM-5.
Introduction
The direct oxidation of lower alkanes to partially oxygenated products remains a major challenge in catalysis. Several strategies have emerged which generally fall into three categories namely: (i) direct high temperature - high pressure activation of alkanes in low yield - high selectivity processes;1–3 (ii) oxidation in strong acidic media using noble metal catalysts to produce alcohol derivatives as esters;4–8 and (iii) activation under moderate conditions using micro/mesoporous catalysts containing transition metal sites and N2O/O2 which operate without catalytic turnover.9–15 To-date whilst many studies have focussed on the direct oxidation of lower alkanes to alcohols and aldehydes, none have proven commercially viable. This has created a situation where energy intensive technologies (e.g. steam reforming of methane to syngas as an integral part of methanol production,16 steam cracking of ethane to ethene followed by hydroxylation to ethanol17) or other technologies (e.g. the BP Cativa Process18) are in global operation.For decades it has been recognised that first row transition metal sites, particularly Fe and Cu, when encapsulated within or supported upon porous materials such as ZSM-5, can perform the oxidation of lower alkanes to alcohols and aldehydes.9–11,14,19–23 These catalysts require N2O (in the case of Fe active sites) or O2 (in the case of Cu active sites) as the oxygen source, but at present these systems do not achieve sufficient catalytic turnover. Our recent work has focussed on using ZSM-5-based materials for the direct low temperature selective oxidation of methane to oxygenated products using hydrogen peroxide as the oxidant.24–26 We reported high intrinsic catalytic activity for ZSM-5 in this system and found that the active site can be attributed to trace impurities of iron, which exist as extra-framework di-iron-μ-oxo-hydroxo complexes or oligomeric iron complexes.24–26 Cu was found to successfully mitigate the oxidation of methanol to formic acid and thereby binary Cu–Fe/ZSM-5 catalysts are capable of giving very high selectivity to methanol.24,26
Usually ion exchange, wet impregnation or sublimation of FeCl3 onto ZSM-5 followed by appropriate heat treatments are used to prepare Fe/ZSM-5 materials for use in alkane activation studies.27,28 Although these methods are well known, there are certain drawbacks to all of these preparation procedures such as the extended time required for metal deposition, difficulty in precisely controlling the iron loading, the presence of spectator ions, and the agglomeration of iron into large oxide particles upon high temperature heat treatments (necessary for activation of the ZSM-5).29 For materials prepared by post-synthesis deposition of Fe (to increase the catalytic activity) there can be multiple types of iron species present in the material and thus the individual contribution of each type of Fe site to the catalysis is difficult to unravel. We have found that chemical vapour impregnation (CVI) is a useful technique for depositing highly dispersed metal species onto a variety of supports and employed this technique to ZSM-5 catalysts in an effort to develop a facile, reproducible and easily controlled metal deposition process.30,31 Recently we reported that Fe and Fe–Cu/ZSM-5 catalysts prepared by CVI are very effective for the aqueous phase oxidation of ethane to acetic acid and ethene using hydrogen peroxide as the oxidant below 70 °C.30 In the current paper we have extended our studies and now report additional characterisation data for these catalysts which show that the majority of the metal sites are isolated. In addition, we demonstrate that heat treatment under a reducing atmosphere, as opposed to an oxidising atmosphere, is highly beneficial and leads to an improved iron dispersion within the zeolite pores. Additionally, we show that these hydrogen treated catalysts have significantly higher alcohol selectivity than their calcined analogues, illustrating the importance of fine-tuning the structure and distribution of the iron species in ZSM-5 and optimising their oxidation state.
Results
Catalyst preparation using chemical vapour impregnation
Vapour deposition strategies can be a very efficient and clean approach for depositing a variety of metals onto oxidic support materials.32 We have used the new approach of chemical vapour impregnation (CVI)30,31 in this current work to prepare our catalyst materials. We selected acetylacetonate salts as the organometallic precursor due to their low cost, ease of handling (i.e. they are solid under ambient conditions, as well as stable in light and air), moderate sublimation temperature (100–150 °C) and the availability of a wide number of metal acetylacetonate variants. Catalysts with different metal loadings and heat treatment conditions were prepared using H-ZMS-5(30) as the support. A summary of metal loadings and heat treatment conditions used are given in Table 1 and the preparation methodology is described in fuller detail in the Experimental section of the ESI.† ICP analysis on samples with nominal iron loadings of 2.5 wt%, 1.1 wt%, 0.4 wt% were found to have actual loadings of 2.4 wt%, 1.02 wt% and 0.41 wt% Fe respectively, which supports the assertion that our method allows the controlled deposition of pre-determined amounts of iron onto H-ZSM-5.| Sample ID | Metal precursor | Nominal metal loading (wt%) | Support | Heat treatment conditions |
|---|---|---|---|---|
| Fe/ZSM-5_#1 | Fe(acac)3 | 1.1 | ZSM-5(30) | Air, 550 °C, 3 h |
| Fe/ZSM-5_#2 | Fe(acac)3 | 2.5 | ZSM-5(30) | Air, 550 °C, 3 h |
| FeCu/ZSM-5_#3 | Fe(acac)3 | 1.25 | ZSM-5(30) | Air, 550 °C, 3 h |
| Cu(acac)2 | 1.25 | |||
| Fe/ZSM-5_#4 | Fe(acac)3 | 1.1 | ZSM-5(30) | 5% H2/Ar, 550 °C, 3 h |
| Fe/ZSM-5_#5 | Fe(acac)3 | 1.1 | ZSM-5(30) | He, 550 °C, 3 h |
| Fe/ZSM-5_#6 | Fe(acac)3 | 1.1 | ZSM-5(30) Pre-treated in air at 550 °C, 3 h | 5% H2/Ar, 550 °C, 3 h |
| Fe/ZSM-5_#7 | Fe(acac)3 | 1.1 | ZSM-5(30) | Air, 550 °C, 3 h then 5% H2/Ar, 550 °C, 3 h |
| Fe/ZSM-5_#8 | Fe(acac)3 | 0.4 | ZSM-5(30) | Air, 550 °C, 3 h |
| Fe/ZSM-5_#9 | Fe(acac)3 | 0.4 | ZSM-5(30) | H2/Ar, 550 °C, 3 h |
| Fe/ZSM-5_#10 | Fe(acac)2 | 0.4 | ZSM-5(30) | Air, 550 °C, 3 h |
| Fe/ZSM-5_#11 | Fe(acac)2 | 0.4 | ZSM-5(30) | H2/Ar, 550 °C, 3 h |
| Fe/ZSM-5_#12 | Fe(acac)3 | 2.5 | ZSM-5(30) | 5% H2/Ar, 550 °C, 3 h |
| FeCu/ZSM-5_#13 | Fe(acac)3 | 0.4 | ZSM-5(30) | Air, 550 °C, 3 h |
| Cu(acac)2 | 0.4 |
Catalysis
We employed our CVI derived ZSM-5(30) based catalysts in the oxidation of methane under mild aqueous conditions using hydrogen peroxide as oxidant (see Experimental section of ESI† for further details). We found that the 1.1 wt% Fe/ZSM-5_#1 material showed high catalytic activity (15.8 mol product kg(cat)−1 h−1) with moderate oxygenate selectivity of 82%, (Table 2 entry 1). A catalyst with higher iron loading, 2.4 wt% Fe/ZSM-5_#2, (Table 2, entry 2) showed a slightly higher catalyst productivity level to the 1.1 wt% Fe catalyst but with a loss of oxygenate selectivity (ca. 10% drop) which may be linked to presence of catalytically active surface iron species that decompose hydrogen peroxide to generate reactive oxygen species that lead to non-selective oxidation. This prompted us to focus our efforts on the materials with lower loadings of iron for the subsequent catalysis studies. For materials that were heat treated in air, it was found that formic acid, a sequential oxidation product derived from methanol, was now the major product. We have previously shown that formic acid is linked to the production of hydroxyl radicals produced in the transformation of methyl hydroperoxide (the primary oxidation product) to methanol.24| Entry | Catalyst | Total productivityb,c mol kg(cat)−1 h−1 | MeOH productivityd mol kg(cat)−1 h−1 | Oxygenate selectivitye (%) | MeOH selectivityf (%) |
|---|---|---|---|---|---|
| a Reaction Conditions: reaction time 0.5 h; reaction temp: 50 °C; solvent: H2O, 10 ml; [H2O2]: 0.5 M; P (CH4): 30 bar; catalyst mass: 0.027 g. b mol(CH3OOH + CH3OH + CH2O + HCOOH + CO2)/time (h) × catalyst mass (kg). c Aqueous phase analysed using 1H-NMR and gas phase analysed using GC-FID. d mol(CH3OOH)/time (h) × catalyst mass (kg). e (CH3OH + CH3OOH + CH2O + HCOOH)/(CH3OH + CH3OOH + CH2O + HCOOH + CO2) × 100. f (CH3OH)/(CH3OH + CH3OOH + CH2O + HCOOH + CO2) × 100. g Reaction time 0.167 h, average of three reactions. h Reaction time 0.167 h. | |||||
| 1 | Fe/ZSM-5_#1 | 15.8 | 1.9 | 82 | 12 |
| 2 | Fe/ZSM-5_#2 | 17.5 | 0.2 | 73.2 | 0.9 |
| 3 | Fe–Cu/ZSM-5_#3 | 11.4 | 9.3 | 82.0 | 77.9 |
| 4 | Fe/ZSM-5_#4 | 15.2 | 4.6 | 80.0 | 30.0 |
| 5 | Fe/ZSM-5_#5 | 16.0 | 5.1 | 80.0 | 32.1 |
| 6 | Fe/ZSM-5_#6 | 16.7 | 6.0 | 84.0 | 36.0 |
| 7 | Fe/ZSM-5_#7 | 15.5 | 2.5 | 76.0 | 16.0 |
| 8g | Fe/ZSM-5_#8 | 24.9, (±2.4) | 3.9, (±0.1) | 93.7, (±0.1) | 16.3, (±0.9) |
| 9g | Fe/ZSM-5_#9 | 26.9, (±1.8) | 9.3, (±0.6) | 90.1, (±5.9) | 35.7, (±0.2) |
| 10h | Fe/ZSM-5_#10 | 8.40 | 1.7 | 94.0 | 20.6 |
| 11h | Fe/ZSM-5_#11 | 12.4 | 4.0 | 91.6 | 32.2 |
The beneficial effect of using Cu as either (i) a homogeneous additive, (ii) a physical mixture with a supported Cu catalyst, or (iii) a bimetallic Fe–Cu/ZSM-5(30) catalyst, on the activity of Fe species in ZSM-5(30) to arrest the formation of formic acid in this reaction has been previously demonstrated.24–26 To ascertain if this situation was also the case with our CVI materials a bimetallic 1.25 wt% Fe-1.25 wt% Cu/ZSM-5_#3 catalyst was prepared and tested. This material (Table 2, entry 3) showed similar trends to those we previously reported in terms of exhibiting high MeOH selectivity as opposed to high formic acid selectivity24–26 (i.e. with no reduction in overall catalytic activity). Although we have previously proposed that the change in product selectivity was due to the ability of Cu to control the production of hydroxyl radicals (as deduced from EPR radical trapping studies24) other groups have proposed that the CuI ion promotes the heterolytic cleavage of the O–O bond of MeOOH to produce MeOH.33 In the case of 1.25 wt% Fe-1.25 wt% Cu/ZSM-5_#3, CuII species are present at the start of the reaction (ESI Fig. S1†) but we note that the presence of H2O2 would allow the formation of CuI species. It is clear from the prior observation that homogeneous copper salts and physical mixtures of an active Fe/ZSM-5 (or Fe-Silicalite-1) catalyst with a catalytically inactive supported Cu catalyst both arrest the formation of formic acid24,25 that the effect is not necessarily due to the formation of a Fe–Cu alloy or intimately mixed Fe and Cu sites in the zeolite. However, for our bimetallic CVI 1.25 wt% Fe-1.25 wt% Cu/ZSM-5_#3 catalyst there is the additional possibility of the formation of alloyed Fe–Cu nanoparticles/clusters and closely spaced cationic Fe sites (substituting for Al3+ in the framework or as extra-framework species) and Cu sites (extra-framework species) in the zeolite channels. Our EXAFS studies showed little evidence of short range Fe–Cu interactions in analogous CVI Fe–Cu/ZSM-5 materials (see Section on EXAFS characterisation) and thus it is highly probable that our CVI materials are behaving in a manner similar to that previously reported for the physical mixtures.24–26
We noted recently that heat treatment of a Cu/ZSM-5(30) catalyst (prepared by ion exchange) in flowing helium prior to reaction resulted in a loss of the beneficial effect of Cu.25,26 It was proposed that a change in valence state of the Cu ions resulted in an alteration in the mechanistic pathways that consequently led to the high formic acid selectivity instead of high methanol selectivity observed with calcined Cu catalysts. Considering that the Fe species in ZSM-5 are also known to undergo auto-reduction when treated in helium at high temperatures,34,35 we investigated the effect of heat treatments under (i) 5% H2/Ar or (ii) He atmospheres on our materials. To investigate the activity of the hydrogen treated materials in the absence of methane we performed a reaction with H2O2 under nitrogen atmosphere (ESI Table S1†). No oxygenated products were detected and only a minor amount of CO2 was detected in the gas phase reaction mixture (ca. 8 micromoles) with a high level of H2O2 decomposition (ca. 96%). These data show that the catalyst cannot produce oxygenates in the absence of reactant gas (methane). When 1.1 wt% Fe/ZSM-5_#4 (5% H2/Ar treated) and 1.1 wt% Fe/ZSM-5_#5 (He treated) catalysts were employed for the oxidation of methane, we observed a marked increase in methanol selectivity at similar overall catalytic productivity, as compared to the 1.1 wt% Fe/ZSM-5_#1 material which was just heat treated in air, (Table 2 entries 4, 5 versus 1).
We considered that other forms of treatments may also affect the catalysis and so we performed a pre-calcination step on ZSM-5(30) at 550 °C, to activate the zeolite, before CVI deposition of FeIII acetylacetonate (Fe(acac)3) followed by heat treatment under 5% H2/Ar at 550 °C. This material denoted as 1.1 wt% Fe/ZSM-5_#6 displayed a markedly higher methanol selectivity (at similar levels of total productivity) than 1.1 wt% Fe/ZSM-5_#1 which was solely heat treated in air, (Table 2, entry 6 versus 1). However, when the as-prepared material which was heat treated in air, was subjected to a second treatment in 5% H2/Ar atmosphere at 550 °C (Fe/ZSM-5_#8), it showed no difference in catalytic productivity or selectivity as compared to 1.1 wt% Fe/ZSM-5_#1, (Table 2, entry 7 versus 1).
Since zeolites with high iron loadings can form surface iron species upon heat treatment, we used samples 0.4 wt% Fe/ZSM-5_#8 and 0.4 wt% Fe/ZSM-5_#9 with lower iron loadings in our XAS studies. These catalysts were also evaluated for the oxidation of methane under initial rate conditions (i.e. 10 min of reaction). The data (Table 2, entry 8 versus 9, each an average of 3 reactions) unambiguously show that heat treatment under flowing 5% H2/Ar also leads to a higher methanol selectivity in the catalysts with lower metal loading. To complete our evaluation of catalytic performance we also prepared analogous materials using FeII acetylacetonate as the metal precursor in place of Fe(acac)3. 1.1 wt% Fe/ZSM-5_#10 and 1.1 wt% Fe/ZSM-5_#11 displayed the same trends as their analogues prepared from Fe(acac)3 (Table 2, entries 10, 11). We noted that some of the precursor was not impregnated into the zeolite and so the iron loading was probably lower than expected. However, the catalysts performed in a similar manner to their counterparts and in this regard a difference in the valence state of iron in the precursor resulted in higher methanol selectivity only if the material was also subjected to a heat treatment in 5% H2/Ar prior to reaction.
We also focussed on the oxidation of ethane since we have recently shown the high catalytic activity of Fe/ZSM-5(30) heat treated in air for this particular reaction.30 Acetic acid and formic acid were the major products, the latter being derived from the catalytic cracking of acetic acid and the oxidation of ethylene formed in-situ from ethane in the reaction.30 Acetic acid can be derived from acetaldehyde (produced from ethyl hydroperoxide), ethanol or ethene (a reaction product that is also oxidised in the system). Moreover we have unambiguously shown that ethanol, produced with low selectivity at 50 °C, is derived directly from ethane since ethyl hydroperoxide (a reaction intermediate) does not decompose to form ethanol over monometallic Fe, Cu or bimetallic Fe–Cu catalysts.30,36,37 Our present catalytic data show that the 1.1 wt% Fe/ZSM-5_#4 (heat treated in 5% H2/Ar) had 92% higher ethanol selectivity than 1.1 wt% Fe/ZSM-5_#1 (heat treated in air) and an accompanying higher methanol selectivity (since methanol is derived from ethanol in this system) when tested in the oxidation of ethane (see Fig. 1a). We have previously shown that ethanol selectivity is heavily influenced by the reaction temperature since ethanol is readily decomposed to acetic acid over Fe/ZSM-5 above 30 °C.30 Thus our finding that higher ethanol selectivity is achieved even when the reaction is performed at 50 °C employing the 5% H2/Ar treated catalyst is significant considering that ethanol is also being oxidised as it is produced at the active site. This suggests the rate of oxidation of ethane to ethanol is higher than the rate of oxidation of ethanol to acetic acid when using the hydrogen treated catalysts. Additionally, we tested the 1.1 wt% Fe/ZSM-5_#4 material under the same slightly altered reaction conditions ([H2O2] of 1 M, 20 ml water, 0.054 g catalyst, 0.0032 mol ethane) used to achieve higher conversion of ethane (ca. 13%) with ca. 12% ethanol selectivity at 30 °C in our previous work.30 We observed under these altered conditions the conversion of ethane was slightly higher (ca. 20%) with a much higher ethanol selectivity of ca. 36% for the 1.1 wt% Fe/ZSM-5_#4 versus 1.1 wt% Fe/ZSM-5_#1 (see Fig. 1b). These data indicate that appreciable conversion of ethane with high ethanol selectivity can be obtained under very mild conditions using our catalysts heat treated in a reducing atmosphere.
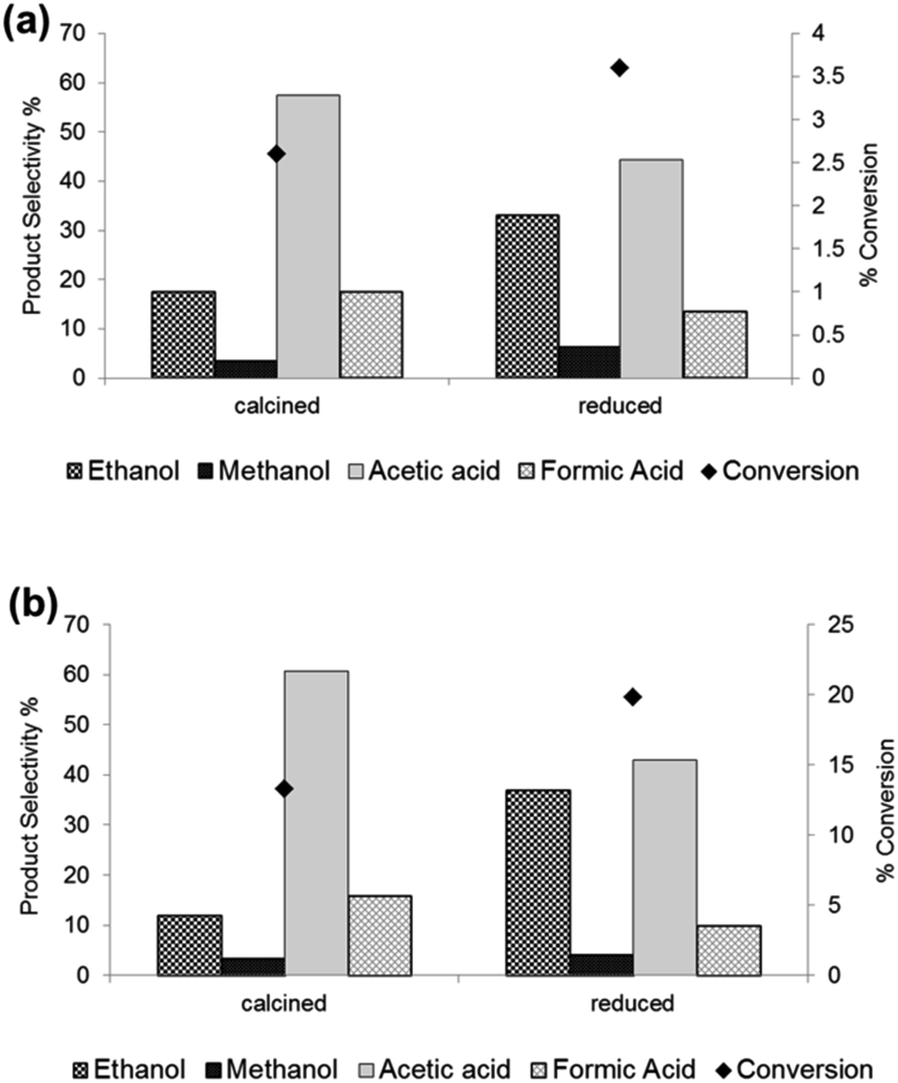 | ||
| Fig. 1 Comparison of the catalytic activity and alcohol selectivity in the oxidation of ethane using H2O2 as oxidant over 1.1 wt% Fe/ZSM-5_#4 heat treated under reducing conditions. Alcohol selectivity refers to ethanol selectivity based on C. (a) Test conditions- 27 mg catalyst; H2O2 : 0.5 M; reaction volume: 10 ml; C2H6 : 0.021 mol; stirring rate: 1500 rpm; reaction temperature: 50 °C, time 0.5 h. (b) Test conditions- 54 mg catalyst; H2O2: 1 M; reaction volume: 20 ml; C2H6 : 0.0032 mol; stirring rate: 1500 rpm; reaction temperature: 30 °C; time: 0.5 h. | ||
Characterisation
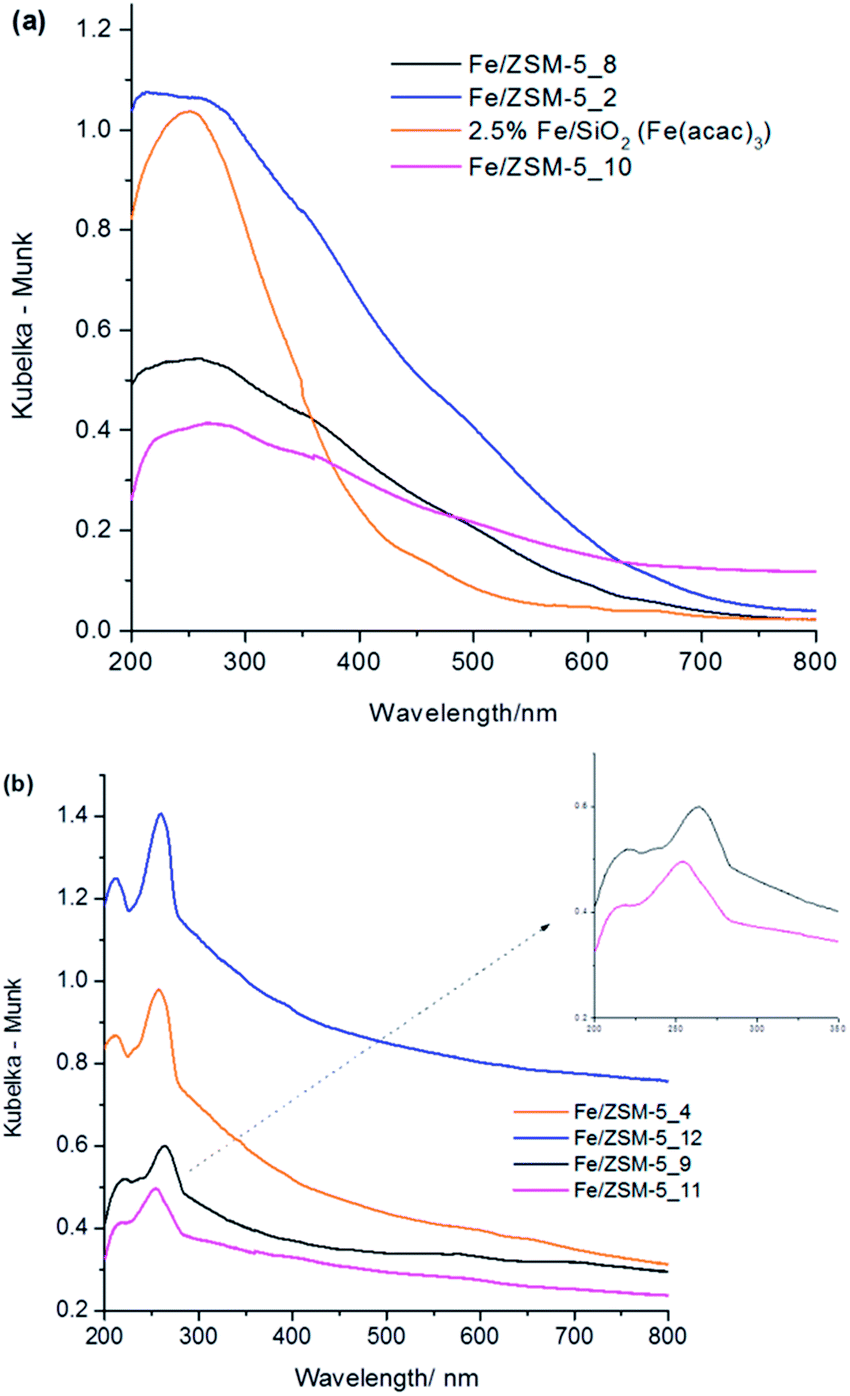 | ||
| Fig. 2 DR UV-vis absorbance spectra of various Fe/ZSM-5 and Fe/SiO2 catalyst materials (a) heat treated at 550 °C in air for 3 h and (b) catalysts heat treated at 550 °C in 5% H2/Ar for 3 h. | ||
 | ||
| Fig. 3 Representative STEM-HAADF images of (a and b) 0.4 wt% Fe/ZSM-5_#8 and (c and d) 2.5 wt% Fe/ZSM-5_#2. Both catalysts were calcined at 550 °C, 3 h in static air. Clustered Fe species on the ZSM-5 surface can be found in both catalysts as they appear brighter in HAADF images (a) and (c). (b) and (d) shows “plain” support areas for comparison. Fig. e and f show representative STEM-HAADF images of 1.1 wt% Fe/ZSM-5_#4 which had been heat treated at 550 °C, 3 h in 5% H2/N2 which are now essentially absent of clustered Fe species. | ||
When the as-prepared 1.1 wt% Fe/ZSM-5(30) is heated in hydrogen at 550 °C, without prior heating in air, the UV-Vis spectra of these materials (Fig. 2b) show marked differences to their analogues formed by heating in air at 550 °C. Clearly resolved bands are found in the range of 200–300 nm in the hydrogen treated samples. The UV-Vis features at 215 nm (major) and 235 nm (minor shoulder peak) for 1.1 wt% Fe/ZSM-5_#4, 0.4 wt% Fe/ZSM-5_#9 and 2.5 wt% Fe/ZSM-5_#12 fall in the region characteristic of isolated FeIII ions in framework positions within MFI structures.35,43 In particular, a band at 215 nm has been previously observed and assigned to FeIII in tetrahedral (Td) coordination within the framework of Fe-silicalite-1 and uncalcined Fe/ZSM-5 prepared by hydrothermal synthesis.41,43,45 Furthermore, it has been noted that a band ca. 235 nm is formed by dehydrating extra-framework FeIII species with octahedral (Oh) coordination in Fe/ZSM-5.34 However, iron complexes with Oh coordination may also show charge transfer bands in this region and hence unambiguous assignment of the precise species responsible for these bands is not possible using UV-Vis spectroscopy as a stand-alone characterisation method.41,43,44 A third distinct band at ca. 260 nm (Fig. 2b) can be assigned to small extra-framework FeIII oxo clusters based on studies of iron in H/ZSM-5 by Hensen et al.42 and in Fe/silicalite-1 by Brodiga et al.43 In the present work, where we have used the CVI preparation method, we note that in the metal precursor, the FeIII centre is octahedrally coordinated to oxygen and decomposition under a reductive atmosphere may induce a different decomposition mechanism as compared to decomposition in the presence of oxygen. We also postulate that the process of calcination of hydrolysed Fe species in Fe/ZSM-5 prepared by sublimation of FeCl3 is also very different from the decomposition of Fe species in our materials under a reducing atmosphere.
We also investigated 0.4 wt% Fe/ZSM-5_#11, which was prepared from Fe(acac)2, using DR UV-Vis spectroscopy (Fig. 2). We anticipated that the difference in precursor properties (i.e. ease of sublimation, propensity to form oligomers, decomposition characteristics) might impose major differences in the catalysts when heat treated in a reducing atmosphere. The UV-Vis spectrum of 0.4 wt% Fe/ZSM-5_#11 was similar to that observed for 0.4 wt% Fe/ZSM-5_#9 (derived from Fe(acac)3) but with a systematic blue-shifted pattern of bands accompanied by a broadening of the major peaks. An expanded view of the region of interest (inset of Fig. 2b) clearly shows that the most intense band at ca. 260 nm is actually composed of at least two peaks centered at 250 nm and 270 nm. Both of these peaks are normally attributed to isolated octahedral FeIII ions present as iron oxo complexes or iron oxide clusters in the ZSM-5 channels when prepared from an FeIII precursor.42 However, in the case of a FeII precursor source, such peaks would most likely involve Fe2+ ions as the material was heat treated under reducing conditions. We also note that the spectral feature corresponding to iron oligomers and nanoscopic surface iron oxide clusters (ca. 300–450 nm) are observable in the case of these materials with lower Fe loadings. It is important to note that the contribution of different extra-framework species to the UV-Vis spectra of our samples varies based on the total iron content and the atmosphere encountered during heat treatment. A characteristic peculiar trend found in the spectra of all the materials heat treated in hydrogen (Fig. 2b) is that although there are no well resolved bands above 300 nm, the absorbance is particularly high in this region (i.e. it does not decrease to zero absorbance). Although this may be taken as being due to a contribution from very small iron oxide clusters within zeolite pores (peak ca. 350 nm) or larger surface iron oxide crystallites (peaks > 450 nm) we assert that other surface sensitive techniques are required to clarify this point. Additionally, all of these materials treated in 5% H2/Ar show similar spectral features indicating the possibility that similar iron species (in differing amounts) are being generated in the catalysts even though the iron loading varies between 0.4 and 2.5 wt%. UV-Vis data presented has provided some information about the location and type of iron species contained in these catalysts, but we additionally employed Scanning Transmission Electron Microscopy (STEM) and X-ray Absorptions Fine Structure Spectroscopy (XAFS) to further probe the local environment of the various iron species present in these materials.
A comparison of Fe K-edge XANES spectra for the various standards used in this work are presented in ESI Fig. S2 and S3.† The main peak in the pre-edge region can be assigned to forbidden quadruple 1s → 3d transitions. As can be seen in the spectrum for Fe(II) acetate, this peak is shifted to a lower energy for FeII compared to FeIII, (ESI Fig S3†). The features around the main edge of the XANES spectrum are associated with 1s → 4p dipole transitions.49 The inherent transition strength for a quadrupole transition is ∼0.1% of the intensity of a dipolar transition. However, due to the large density of states of the 3d-band, they appear as small peaks (∼1–3%) in most octahedral transition metal XANES spectra.50 When the co-ordination geometry is switched to tetrahedral (or to a more distorted octahedral) there is mixing between the metal 3d and 4p states, allowing for a direct dipole transition into the 4p character of the 3d band. This results in a much more intense pre-edge feature as shown in ESI Fig. S2.† The pre-edge peak can be broadened for octahedral systems by what are termed as non-local transitions to an inter-site hybrid.51 The broadening exhibited by α-Fe2O3 (ESI Fig. S3†) is a result of the promotion of a 1s electron to 3d states of the next Fe neighbour. This is facilitated by hybridisation of 4p states of the absorbing atom with 3d states of the next Fe atom through O 2p states. As there is hybridisation of the 4p and 3d orbitals, the transition is partially dipolar in nature and can appear more intense than the normal quadrupolar pre-edge excitations.
We used a low iron loaded material (0.4 wt% Fe) for studies on our catalytic system in order to minimise the extent of contribution to the spectra from iron oxide particles on the external surface of the zeolite crystals which commonly are formed when Fe/ZSM-5 undergoes high temperature treatments in air. Notably in the case of the reduced catalyst, we could not detect surface iron oxides (FexOy clusters or large Fe2O3 particles) on the 1.1 wt% Fe/ZSM-5_#4 sample by HAADF-STEM (Fig. 3). Fig. 4a shows the XANES comparison of the 0.4 wt% Fe/ZSM-5_#8 (calcined) and 0.4 wt% Fe/ZSM-5_#9 (reduced) sample with α-Fe2O3 and Fe(II) acetate as reference materials, and the corresponding pre-edge region is enlarged in Fig. 4b. In each catalyst sample the most intense pre-edge feature observed corresponds to that occurring in the octahedral α-Fe2O3 standard, confirming that the majority (if not all) Fe species in each catalyst sample are in an octahedral environment. In addition, there are several indications that there is some FeII present namely (i) an increase in absorption around 7112 eV (corresponding to the FeII 1s → 3d transition),49 (ii) a reduction in the peak at 7114 eV (corresponding to the FeIII 1s → 3d transition) and (iii) a shift of the main edge to a lower energy, all in agreement with previous studies.34 It can be seen that both 0.4 wt% Fe/ZSM-5_#8 (calcined) and 0.4 wt% Fe/ZMS-5_#9 (reduced) have some Fe2+ character, with this being more pronounced in the latter sample which was heat treated in hydrogen. A more intense and slight broadening of the peak at 7114 eV for the calcined sample may also be a sign of an inter-site hybrid and the presence of some Fe–O–Fe interactions.
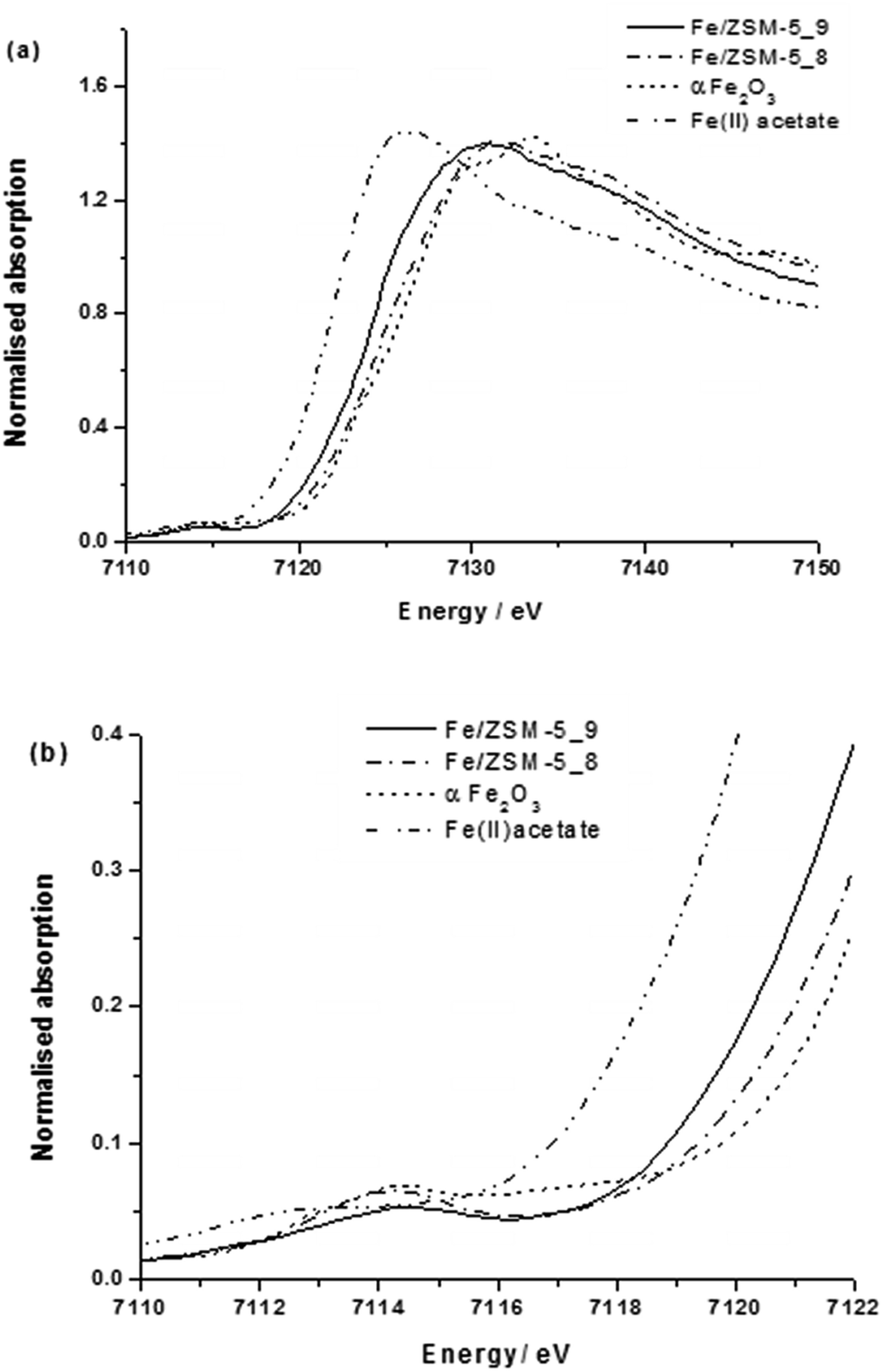 | ||
| Fig. 4 (a) XANES spectra and (b) expanded pre-edge region from various 0.4 wt% Fe/ZSM-5(30) CVI samples. | ||
The corresponding EXAFS spectra of these materials are shown in Fig. 5 with the generated fitting parameters reported in Table 3. The EXAFS data highlight the differences in the local structure of the Fe sites and show that for 0.4 wt% Fe/ZSM-5_#9 (reduced catalyst), the Fe is present with a primary coordination sphere comprising of oxygen. To generate a satisfactory fit, a two shell model of Fe–O distances separated by ∼0.2 Å was included. The absence of any further scattering processes (e.g. Fe–Al shell) is supported by the absence of any other significant peaks in the radial distribution plot (Fig. 5a, right) beyond that associated with the primary oxygen coordination sphere, this is also evidenced by the χ plot (Fig. 5a left), which is consistent with a single primary coordination sphere of low atomic mass (z) neighbours. The major difference observed in the calcined 0.4 wt% Fe/ZSM-5_#8 catalyst is the presence of an Fe–Fe shell, which is most likely a consequence of the presence of oligonuclear extra-framework species or FexOy clusters and surface iron containing species, in agreement with our spectroscopic data from DR UV-Vis and HAADF-STEM analysis in Fig. 2 and 3. The Fe–Fe shell can be seen as an additional peak in the radial distribution plot of the EXAFS data (Fig. 5b, right), which has the effect of dampening the amplitude of the high k oscillations in the χ plot due to the out-of-phase behaviour of the Fe–Fe and Fe–O scattering paths (Fig. 5b, left).
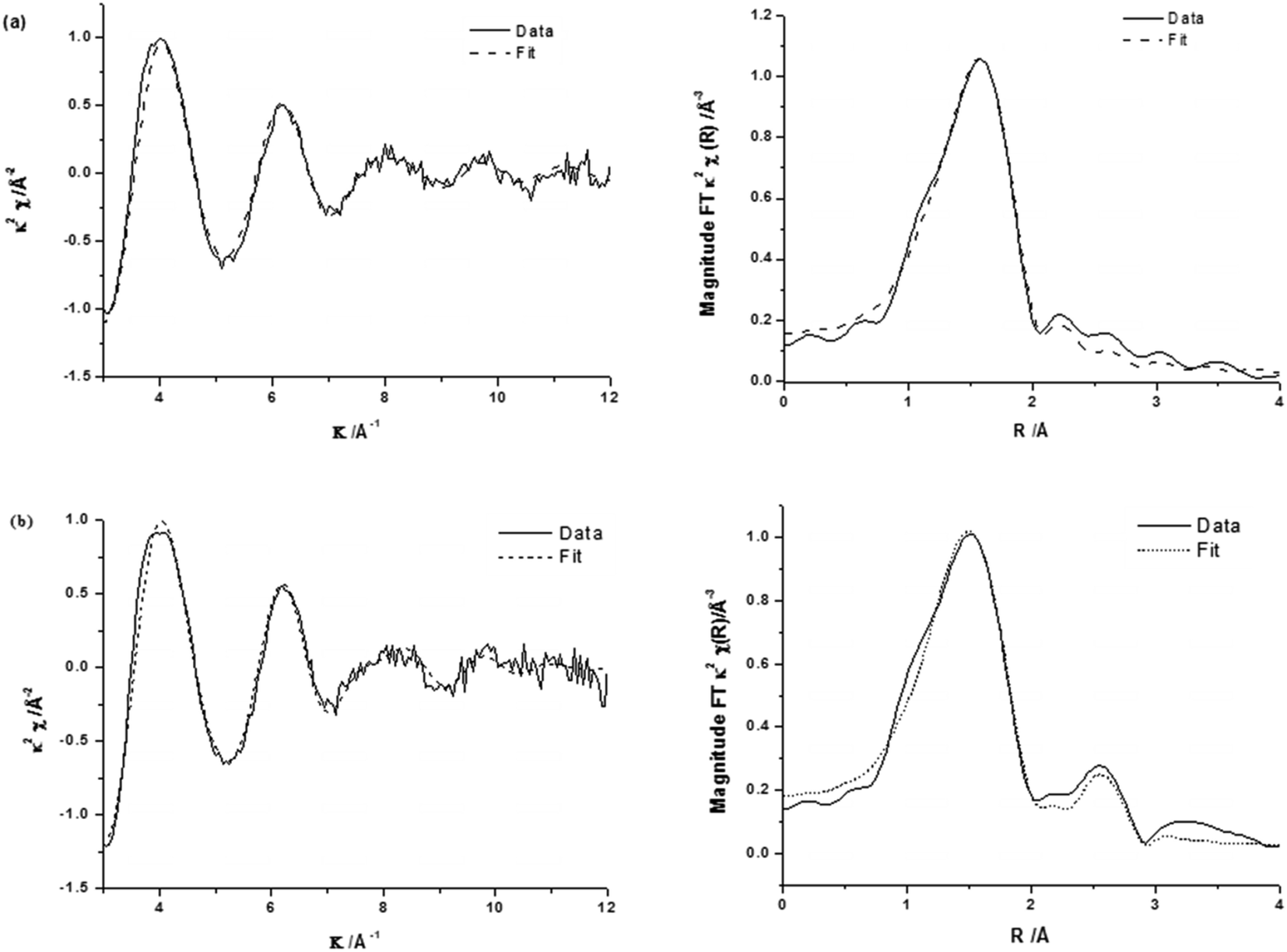 | ||
| Fig. 5 k 2 experimental χ data and fit (left) along with associated Fourier transform (right) for (a) 0.4 wt% Fe/ZSM-5_#9 and (b) 0.4 wt% Fe/ZSM-5_#8. | ||
| Catalyst | Treatment | Abs Sc | N | R Å−1 | 2σ2/Å2 | E f /eV | R factor |
|---|---|---|---|---|---|---|---|
| a Fitting parameters: S02 = 0.73 as deduced using an Fe(PO4)·2H2O standard; Fit range 2 < k < 11, 1 < R < 3; # of independent points = 10. b Fitting parameters: S02 = 0.73 as deduced using an Fe(PO4)·2H2O standard; Fit range 2 < k < 10, 1 < R < 3; # of independent points = 8. c Fitting parameters: S02 = 0.73 as deduced using an Fe(PO4)·2H2O standard; Fit range 2 < k < 11, 1 < R < 3; # of independent points = 10. | |||||||
| Fe/ZSM-5_#8a | 550 °C, Air | Fe–O | 2.5 (9) | 1.95 (3) | 0.005 (3) | −1 (1) | 0.003 |
| Fe–O | 3.5 (9) | 2.08 (2) | 0.005 (3) | ||||
| Fe –Fe | 1.1 (3) | 3.04 (3) | 0.009 (4) | ||||
| Fe/ZSM-5_#9b | 550 °C, H2/Ar | Fe–O | 1.7 (7) | 1.95 (1) | 0.005 (4) | 0 (1) | 0.003 |
| Fe–O | 3.9 (9) | 2.09 (1) | 0.004 (3) | ||||
| Fe–Cu/ZSM-5_#13c | 550 °C, Air | Fe–O | 2.1 (6) | 1.93 (2) | 0.004 (3) | 0 (1) | 0.003 |
| Fe–O | 3.4 (7) | 2.08 (2) | 0.004 (2) | ||||
| Fe –Fe | 0.8 (4) | 3.04 (5) | 0.010 (6) | ||||
XPS measurements of the air-treated and hydrogen-reduced catalyst (Fig. S4†) also support our XAFS data as there is a difference between the iron detected in both catalysts even though both show a +3 oxidation state. When reduced under hydrogen the Fe 2p3/2 peak (711.9 eV with satellite at 720 eV) is characteristic of isolated iron sites such as Fe(O)OH whereas for the calcined material the iron sites are characteristic of small α-Fe2O3 particles/films (Fe 2p3/2 peak at 711.3 eV and 719 eV).52 These data strongly support our XASF data which shows that in the reduced material the iron is found as isolated iron oxo clusters. We note that XPS techniques would not be able to detect/quantify very small fractions of Fe2+ in a mostly Fe3+ sample and Fe located deep within the zeolite crystals cannot be probed by XPS and thus further characterisation using XPS was not performed.
The XAFS data (Fig. 6 and 7 and Table 3, last row) for 0.4 wt% Fe 0.4 wt% Cu/ZSM-5_#13 show that the local Fe structure in the bimetallic Fe–Cu catalyst and monometallic Fe catalysts are very similar when heat treated in air, with the only major difference being the presence of more Fe2+ in the bimetallic sample. This is evidenced by the main edge position being shifted to a lower energy for the Fe–Cu sample (0.4 wt% Fe-0.4 wt% Cu/ZSM-5_#13) as compared to the calcined Fe-only material (0.4 wt% Fe/ZSM-5_#8). Moreover, Fe and Cu have significantly different back-scattering amplitudes, thus Fe–Fe and Fe–Cu scattering processes are easily distinguishable by EXAFS. This indicates the very low probability of Fe–Cu interactions in this catalyst and supports that hypothesis that no bimetallic alloyed nanoparticles are present in 0.4 wt% Fe-0.4 wt% Cu/ZSM-5_#13.
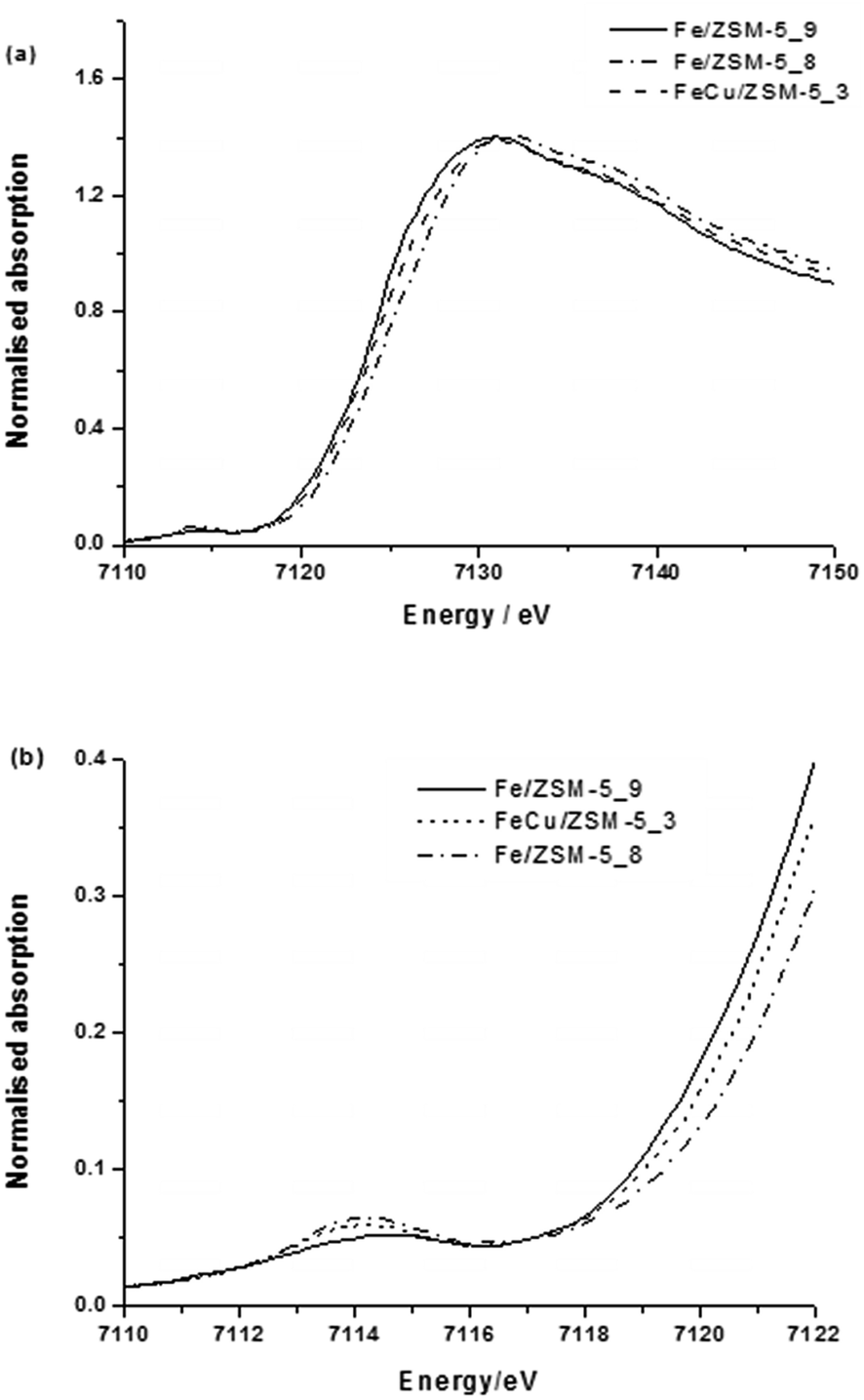 | ||
| Fig. 6 (a) XANES spectra and (b) expanded pre-edge region of XANES spectra of 0.4 wt% Fe-0.4 wt% Cu/ZSM-5_#13 CVI samples. | ||
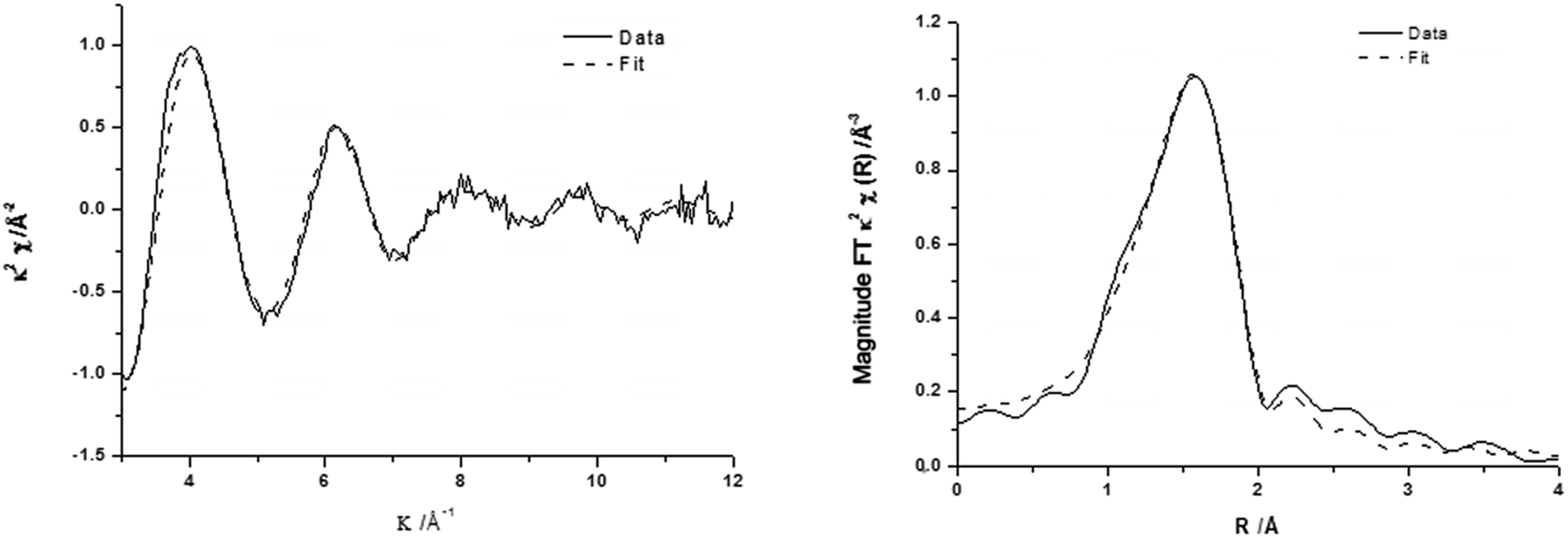 | ||
| Fig. 7 k 2 experimental χ data and fit (left) along with associated Fourier transform (right) for calcined 0.4 wt% Fe-0.4 wt% Cu/ZSM-5_#13 prepared by CVI. | ||
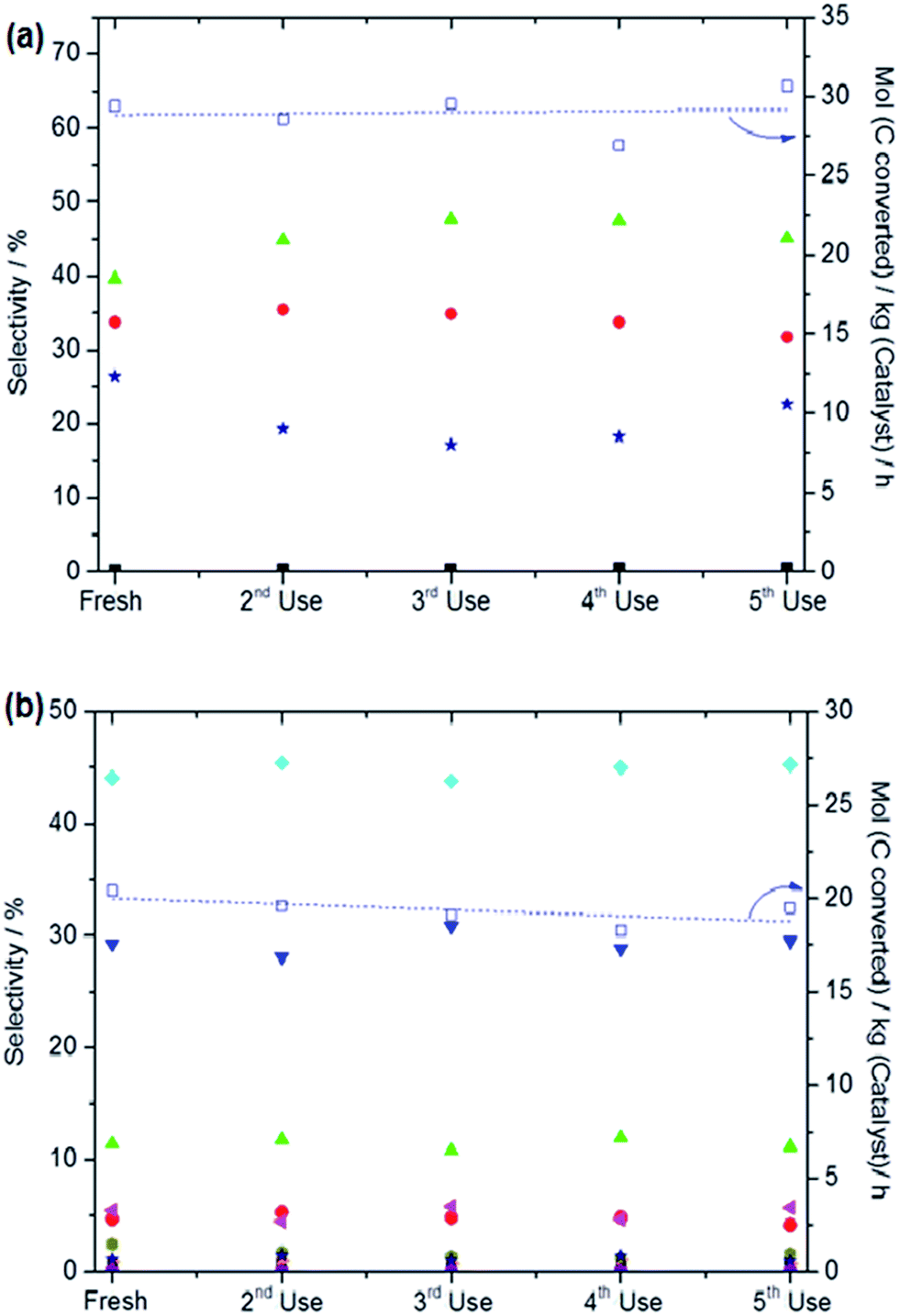 | ||
| Fig. 8 Catalytic performance of 1.1 wt% Fe/ZSM-5_#4 employed in the oxidation of (a) methane and (b) ethane using H2O2 as oxidant over five (5) reuse cycles. Test conditions for (a) 27 mg catalyst; reaction volume: 10 ml; [H2O2]: 0.5 M; reaction temperature: 50 °C; time: 0.25 h; stirring rate: 1500 rpm, P (CH4) : 30 bar. Test conditions for (b) 54 mg catalyst; reaction volume: 20 ml; [H2O2]: 1 M; reaction temperature: 30 °C, time: 0.5 h, stirring rate: 1500 rpm; P (C2H6): 5 bar. | ||
We collected the DR-UV-vis spectra for the hydrogen treated catalysts used for one (1) and five (5) cycles in the oxidation of both methane and ethane, Fig. 9. There are some spectral changes in the used materials compared to the catalyst before exposure to H2O2 and CH4. After one reaction cycle with methane or ethane the Fe bands are less intense but adopt the same shape and position as the unused catalyst. After 5 reaction cycles a reduction in area of the band ca. 260 nm and broadening/increased intensity of the band ca. 215 nm (probably 2 bands at 215 nm and 230 nm) is observed. One explanation would be the conversion of some isolated iron species into new clustered species with different coordination sphere and/or different oxidation state. One would expect that in a highly oxidising aqueous environment the structure and nuclearity of the iron species would be altered. If the catalyst was stirred in water, under nitrogen atmosphere, no change is observed in the UV-Vis spectrum but there is a clear change, in line with that observed in the actual reaction, if the catalyst is stirred in aqueous H2O2 (ESI Fig. S5†). On comparing the DR-UV-vis spectrum of the catalyst used in one cycle with methane/ethane and when stirred solely in aqueous H2O2 it is apparent that the former retains more features of the unused material. However, these changes show little effect on the overall catalytic activity and methanol/ethanol selectivity and this is probably due to the nature of the redox reaction induced by H2O2, i.e. Fe may be changing oxidation state and coordination reversibly during the reaction mechanism. Additional studies for longer reaction time under continuous flow conditions are necessary to further investigate the long term stability of the materials and determine treatments are necessary to mitigate any loss of catalytic activity or product selectivity.
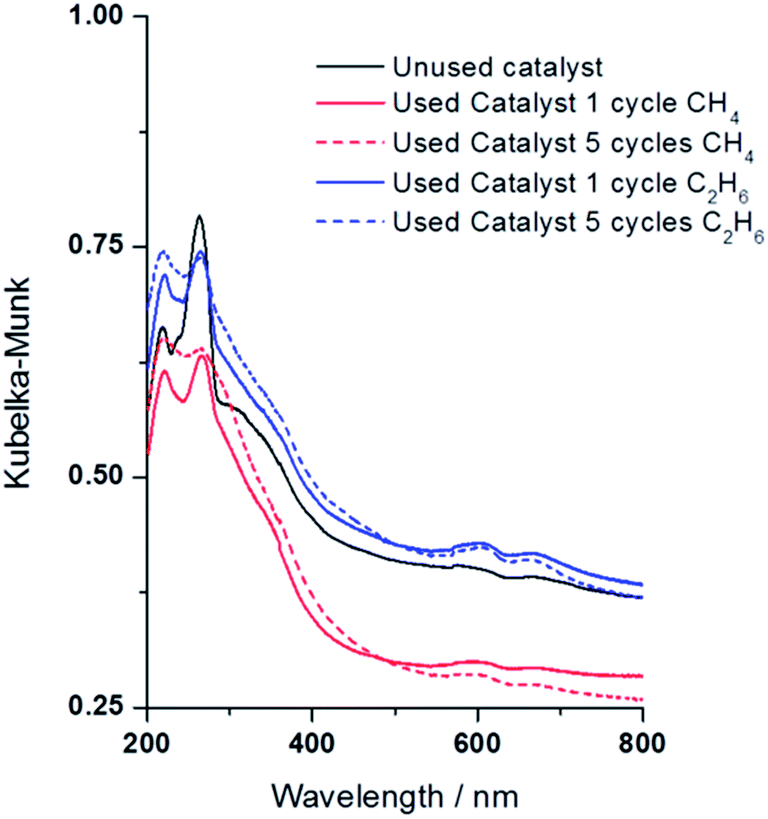 | ||
| Fig. 9 DR UV-vis spectra of 1.1 wt% Fe/ZSM-5_#4 employed in the oxidation of methane and ethane measured after one (1) and five (5) reaction cycles using H2O2 as oxidant. Test conditions as in Fig. 8, catalysts analysed at room temperature after air drying overnight. | ||
Discussion
In the preparation of metal loaded zeolites, post-synthesis deposition of metals using solid or liquid phase methods has been extensively studied. For Fe in Fe/ZSM-5(30), studies have focussed on the elucidation of the catalytically active Fe species and it has been largely assumed that only the iron species found within the ZSM-5 pores are involved in the reaction mechanism. This may be the case where N2O is used as the oxidant forming α-oxygen (in high temperature non-aqueous systems) which is postulated to be the active oxygen species that performs methane oxidation producing methanol (after hydrolysis). In this study, H2O2 is employed as the oxidant under mild aqueous conditions where it has been previously demonstrated that reaction pathways leading to deeper oxidation products (i.e. formic acid and CO2) are in operation. Though we have proposed that the active site in ZSM-5(30) is similar to the di-iron-μ-oxo-hydroxo cluster proposed by other researchers, the question of which iron species in Fe loaded ZSM-5 comprise the active site(s) becomes more complex due to the possibility of speciation within the zeolite pores and on the external zeolite surface as the total iron content is increased.23 We note that in the preparation of iron loaded ZSM-5 by post-synthesis deposition methods considerable amount of surface iron species (iron oxides) are found after calcination treatment.9,53–55Using the CVI methodology we attempted to prepare FeZSM-5 in which the iron species were exclusively within the pores of the zeolite whilst circumventing the time consuming process of hydrothermal synthesis. When our materials were heat treated in air the UV-vis, STEM and XAFS analyses all suggested that a significant amount of Fe was found as iron oxides on the external ZSM-5 surface or within the zeolite pores. This may be expected based on the known ability of iron initially located at cationic exchange sites within zeolite pores to migrate and form external iron oxides upon thermal treatment in air.45 Heat treating Fe loaded ZSM-5 in a reducing atmosphere without prior air treatment has not previously been employed in the preparation of active oxidation catalysts. However, heat treatment in air (first step) followed by helium or hydrogen (second step) for catalysts prepared from FeCl3 by CVD34 or FeCl2 by ion exchange44 has been used to study the reducibility and structure of iron species in ZSM-5 materials. Notably, previous studies showed that “reduced iron species” in Fe/ZSM-5 features an oxygen vacancy created by loss of a bridging ‘O’ from [OH–Fe–O–Fe–OH]2+ core, with the core being formed in the air treatment step, and Fe remains in the +3 oxidation state.34,48 Furthermore, these species could be easily re-oxidised.34,48 Those studies focussed on elucidating the possibility of iron–oxygen clusters in the zeolite donating (i.e. losing) oxygen in redox processes34,48 although no accompanying catalytic studies involving methane were attempted. As shown by Choi and co-workers using XAS56 the structure of Fe in carefully prepared Fe-ZSM-5 shows little change with varying Fe/Al ratio when treated under reducing atmospheres of He or CO and is best described as Z−[FeO2]+ or Z−[Fe(OH)2]+, where Z− is a charge exchange site in the zeolite. Satisfactory fitting of the data could be done with two Fe–O shells but longer range Fe–(OH2) interactions are also possible. Pirngruber and co-workers44 also proposed that the Z−[Fe (OH)2]+ model is appropriate for describing the structure of Fe in calcined 0.4 wt% Fe-ZSM-5(24), though the usual preference for Fe as binuclear iron-oxo-hydroxy clusters in higher loaded materials was found to be inconsistent with several other data. In all of these studies the structure models placed Fe in tetrahedral coordination when considering isolated iron ions.44,56 Traditionally EXAFS fitting has been performed using an Fe–Al (longer range) and 1–3 Fe–O (shorter range) shells in fitting models.
With our materials there is a clear difference in the catalyst when heat treated under hydrogen as shown in the XAFS studies. The absence of an Fe–Fe shell and the higher FeII character in the case of the hydrogen treated catalyst (0.4 wt% Fe/ZSM-5_#9) are the major differences observed when compared to the analogous sample heat treated in air, 0.4 wt% Fe/ZSM-5_#8. The absence of any Fe–Fe contributions in the EXAFS fit of the reduced material confirms that the majority of Fe species (both FeII and FeIII) are present in an isolated extra-framework octahedral environment. However, we cannot rule out the presence of small contributions from other species such as the dimeric iron oxo-hydroxo species as previously reported by Battison et al.48 We also highlight that an Fe–Al shell was not necessary, nor appropriate, in fitting the data and therefore we consider that our iron-oxo species do not have appreciable interaction with framework Al3+ other extra-framework Al species, at distances normally reported in EXFAS fitting. This finding is compounded by the definite octahedral coordination of Fe3+ found in our studies, as opposed to tetrahedral Fe3+ used in earlier models. Thus our new isolated iron species are unlike those previously postulated to have been created by the removal of oxygen from binuclear iron-oxo-hydroxo cores and also those thought to be stabilised at extra-framework cationic exchange sites through Al–O–Fe or Si–O–Fe interactions. We consider this to be a positive advance in preparing new iron species in ZSM-5. Based on careful review of the literature and our own data set we cannot provide a definitive model of the active site but can confirm that octahedral isolated Fe3+/Fe2+ ions with a primary coordination sphere of oxygen, which may be provided by OH− and OH2 ligands, are to be found in our materials. We consider that the hydrated state of the Fe site is due to the storage of the material under ambient conditions and since the catalytic reaction is performed in aqueous environment, unlike all previous studies aimed at linking the structure of Fe to the catalytic activity for non-aqueous high temperature reactions, it is equally important that the proposed active site be stable in aqueous conditions. In previous works water was found to be detrimental to the catalytic activity for N2O decomposition.44,57
Additionally, DR UV-vis analysis of samples heat treated in hydrogen show that irrespective of iron loading the Fe-speciation in these materials are similar in nature, although the fractional contribution of the different species may vary as the iron loading is altered. In particular, we observe bands in the spectra of hydrogen treated materials (Fig. 2b) that support the XAFS data which points to the presence of isolated extra-framework octahedral iron-oxo species and allow us to draw similar conclusions to Choi and co-workers56 concerning the structure of isolated iron sites in ZSM-5 as the iron loading varies. The bands correlating to such species are well resolved, which is not the case for materials treated in air. To the best of our knowledge, this is the first report of such well resolved bands in the UV-vis spectrum of heat treated ZSM-5 materials that contain iron loaded by post-synthesis deposition techniques. This suggests that the decomposition route of Fe(acac)3 and Fe(acac)2 deposited onto ZSM-5 changes based on the atmosphere used in heat treatment. The availability of oxygen would support the formation of iron oxides and larger iron clusters in the air treated samples, whereas the lack of external oxygen (as opposed to the internal oxygen contained in the ligands of the metal precursor) leads to the formation of iron-oxo clusters within the zeolite pores. Since this is true, even at higher iron loadings, we can propose a correlation between the data obtained from XAFS, DR UV-vis, STEM and our catalysis studies.
The higher methanol selectivity observed for all hydrogen treated catalysts used in this study can be linked to the presence of isolated iron-oxo clusters (and possibly oligomers). This is a significant finding, as we previously found that only the inclusion of Cu2+ in the reaction mixture resulted in control of the methanol selectivity.24,26 It could be argued that the high methanol selectivity may also be linked to an increased level of Fe2+ in the hydrogen treated samples, since our XAFS studies demonstrate that the heat treatment under reducing atmosphere leads to a higher proportion of Fe2+ species in the monometallic catalyst. Notably, the active site in ZSM-5 proposed in other studies, i.e. diiron-μ-oxo-hydroxo species, would most likely be disrupted by heat treatment under hydrogen at 550 °C and also undergo facile re-oxidation.34,48 Since our catalytic and characterisation studies were performed on catalysts stored under ambient conditions prior to use, one would anticipate that if extra-framework diiron-μ-oxo-hydroxo species became dehydrated or reduced during the heat treatment under hydrogen, such species would already be rehydrated and re-oxidised before use in our studies. Our catalyst reuse data also shows that the hydrogen treated materials maintained the catalyst productivity and alcohol selectivity (Fig. 8) in the oxidation of both methane and ethane over five reaction cycles without heating the used catalysts in a reducing atmosphere in-between runs (i.e. the used catalyst was dried in air overnight). This is an important finding which shows that the effect of heat treatment under a reducing atmosphere is not a transient phenomenon. Therefore we do not consider that there is strong evidence for a direct correlation between the beneficial alcohol selectivity effect observed in our catalytic studies using hydrogen treated Fe/ZSM-5 and electronic or configuration changes that have been shown to occur in these types of materials when prepared by traditional techniques. Also, due to the absence of larger surface oxides (as confirmed by HAADF-STEM studies) in the samples heat treated under reducing conditions, we propose that the bulky iron oxides are not involved at all in the catalysis using the H2/Ar treated materials.
Furthermore, in the oxidation of methane we found that the 1.1 wt% Fe/ZSM-5_#7 sample did not show an increased methanol selectivity as compared with all other materials that had been solely heat treated in hydrogen. In this particular sample case, heat treatment of the ‘as-prepared’ material in air was followed by heat treatment under hydrogen at 550 °C. According to previous H2-TPR studies of air treated Fe/ZSM-5 (prepared from FeCl3, incipient wetness and ion exchange methods) only the local environment of iron-oxo-hydroxo dimers/oligomers in non-framework sites, and not larger iron aggregates on the zeolite surface, would be disrupted by hydrogen treatment at 550 °C.29,35,53 One would anticipate that only the involvement of these diiron-oxo-hydroxo type species in the catalytic mechanism (assuming they are the active site) would be affected in 1.1 wt% Fe/ZSM-5_#7. Therefore, the absence of a positive effect on methanol selectivity for this material suggests that diiron-oxo-hydroxo type species are not necessarily responsible for the formation of methanol. The catalytic data (Table 2, entry 7) clearly shows that once the active iron species have been formed from the Fe(acac)3 (by heat treatment in air) then hydrogen treatment at 550 °C has little effect on the methanol selectivity. However, if only the ZSM-5 support material is activated by heat treatment in air, and then the metal precursor is absorbed onto the zeolite after such a pre-calcination step, the same iron species as other hydrogen treated catalysts can be prepared after a final heat treatment in a reducing atmosphere and the corresponding catalytic benefit of high methanol selectivity is observed (Table 2, entry 6). Together these two pieces of data validate our hypothesis that different iron species are formed when Fe(acac)3 loaded onto ZSM-5 is decomposed under an oxidising versus reducing atmosphere and that these species are not fully inter-convertible. The observed benefit of a reductive heat treatment step on Fe/ZSM-5 is dependent on the absence of external oxygen in the system prior to high temperature heat treatment.
We previously proposed that in the oxidation of methane using H-ZSM-5 methanol is derived from methyl hydroperoxide with the release of hydroxyl radicals, which are subsequently responsible for the formation of deeper oxidation products.24 We consider that in the present case, the observed effect related to catalyst treatment under hydrogen may be due to (i) a lower methanol decomposition to formic acid, (ii) a different pathway from methyl hydroperoxide to methanol or (iii) an additional pathway to produce methanol directly. The role of increased Fe2+ may be important in the latter scenario as ferryl type species can be accessed from the interaction of Fe2+ sites and hydrogen peroxide, in which case methanol could possibly be produced directly from this type of reactive oxygen species. In depth mechanistic studies using in-situ techniques may unravel the complexity of these catalytic pathways but one can also gather insights from catalytic data in the oxidation of ethane. Ethanol is not stable over Fe/ZSM-5 catalysts in the presence of H2O2 and readily decomposes to methanol, acetaldehyde and acetic acid.58 The importance of our previous findings that ethanol and ethylene were directly produced from ethane under mild aqueous reaction conditions30 has been linked to other computational studies which showed that [M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]+ species could directly produce either ethanol or ethylene from ethane.59,60 In the current case, isolated cationic iron sites should give rise to higher ethanol selectivity when H2O2 is used as oxidant since [FeO]+ species may be easily accessed. Since we have already shown there is a direct route to ethanol in the ethane oxidation reaction, we have additional data (Fig. 1) that further indicates that the iron species in the H2 reduced 1.1 wt% Fe/ZSM-5_#4 perform the direct transformation of ethane to ethanol at levels above that possible with the calcined 1.1 wt% Fe/ZSM-5_#1 and our observations are not merely due to a change in alcohol stability.§
O]+ species could directly produce either ethanol or ethylene from ethane.59,60 In the current case, isolated cationic iron sites should give rise to higher ethanol selectivity when H2O2 is used as oxidant since [FeO]+ species may be easily accessed. Since we have already shown there is a direct route to ethanol in the ethane oxidation reaction, we have additional data (Fig. 1) that further indicates that the iron species in the H2 reduced 1.1 wt% Fe/ZSM-5_#4 perform the direct transformation of ethane to ethanol at levels above that possible with the calcined 1.1 wt% Fe/ZSM-5_#1 and our observations are not merely due to a change in alcohol stability.§
We noted in previous work that a bimetallic Fe–Cu/ZSM-5 catalyst did not display higher alcohol selectivity when applied to ethane oxidation, as was observed in the oxidation of methane, but instead increased the production of ethene from ethane.30 Controlling of ethanol selectivity was not possible with these Fe–Cu/ZSM-5 catalysts heat treated in air thus our finding that treatment of the Fe/ZSM-5 catalyst in hydrogen results in higher ethanol selectivity is a positive practical step towards controlling the product distribution. In an effort to extend our characterisation studies, we are currently performing high level in situ characterisation of the iron species under reaction conditions to investigate the origin of the inserted oxygen and which iron species are involved in the mechanism of oxygen insertion into the C–H bond.
Conclusions
Catalyst design methodologies have been employed which afford another route to tuning alcohol selectivity in the direct oxidation of both methane and ethane under mild conditions. In particular, heat treatment of Fe/ZSM-5 materials prepared by post-synthesis chemical vapour impregnation (CVI) under a reducing atmosphere produces highly dispersed iron species. These species are mostly in the form of isolated octahedral extra-framework iron oxo clusters (and possibly oligomeric species) in the zeolite channels and the catalysts contain higher amounts of FeII than their air treated analogues. We propose the higher alcohol selectivity (measured at similar levels of catalytic productivity) observed in our catalytic studies of the hydrogen/helium versus air treated materials is linked to the presence of these iron-oxo clusters and/or FeII in the catalysts. The importance of the sequence of heat treatment conditions (i.e. in air only, in hydrogen only or in air followed by hydrogen) in creating a beneficial effect on alcohol selectivity suggests that the mechanism of formation of the active iron sites differs in our methodology as compared to others previously reported. The observation of high ethanol selectivity in the oxidation of ethane is undoubtedly linked to a direct route from ethane to ethanol, as opposed to routes through alkyl peroxy intermediates. This strongly suggests the possibility that there is also a direct route from methane to methanol in our system which is an important hypothesis that requires further investigation. The identification of well-defined iron species in Fe/ZSM-5 plays a pivotal role in investigating the contribution of the various iron species in Fe/ZSM-5 to the catalytic activity and product distribution in alkane oxidation reactions. The fact that additional metals (such as Cu) are not required to afford a level of product selectivity control is beneficial in elucidating which are the catalytically active forms of iron (and the associated reactive oxygen species they produce from H2O2) as it renders the system less complex. Our studies expand the area of Fe/ZSM-5 catalysis relating to alkane activation as we move towards more effective catalysts for low temperature selective alkane oxidation.Acknowledgements
We acknowledge EPSRC for funding (EP/I019693/1, EP/K014714/1) and Diamond Light Source for provision of beam time (SP8071-1) The Research Campus Harwell are also acknowledged for use of facilities and support of their staff.References
- N. R. Hunter, H. D. Gesser, L. A. Morton, P. S. Yarlagadda and D. P. C. Fung, Appl. Catal., 1990, 57, 45–54 CrossRef CAS.
- N. R. Foster, Appl. Catal., 1985, 19, 1–11 CrossRef CAS.
- H. D. Gesser, N. R. Hunter and C. B. Prakash, Chem. Rev, 1985, 85, 235–244 CrossRef CAS.
- A. Sen, Acc. Chem. Res., 1998, 31, 550–557 CrossRef CAS.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Russ. Chem. Rev., 1987, 56, 442–464 CrossRef PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- C. Jones, D. J. Taube, V. R. Ziatdinov, R. A. Periana, R. J. Nielsen, J. Oxgaard and W. A. Goddard, Angew. Chem., 2004, 116, 4726–4729 CrossRef.
- G. I. Panov, A. K. Uriarte, M. A. Rodkin and V. I. Sobolev, Catal. Today, 1998, 41, 365–385 CrossRef CAS.
- P. P. Knops-Gerrits and W. A. G. III, J. Mol. Catal. A: Chem., 2001, 166, 135–145 CrossRef CAS.
- B. R. Wood, J. A. Reimer, A. T. Bell, M. T. Janicke and K. C. Ott, J. Catal., 2004, 225, 300–306 CrossRef CAS PubMed.
- K. A. Dubkov, V. I. Sobolev, E. P. Talsi, M. A. Rodkin, N. H. Watkins, A. A. Sheinman and G. I. Panov, J. Mol. Catal. A: Chem., 1997, 123, 155–161 CrossRef CAS.
- N. S. Ovanesyan, A. A. Shteinman, K. A. Dubkov, V. I. Sobolev and G. I. Panov, Kinet. Catal., 1998, 39, 792–797 CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394–1395 CrossRef CAS PubMed.
- N. V. Beznis, A. N. C. v. Laak, B. M. Weckhuysen and J. H. Bitter, Microporous Mesoporous Mater., 2011, 138, 176–183 CrossRef CAS PubMed.
- M. C. Alvarez-Galvan, N. Mota, M. Ojeda, S. Rojas, R. M. Navarro and J. L. G. Fierro, Catal. Today, 2011, 171, 15–23 CrossRef CAS PubMed.
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2012, 398, 1–17 CrossRef PubMed.
- H. Jones, Platin. Met. Rev., 2000, 44, 94–105 Search PubMed.
- G. I. Panov, G. A. Sheveleva, A. S. Kharitonov, V. N. Romannikov and L. A. Vostrikova, Appl. Catal., 1992, 83, 31–36 CrossRef.
- P. Knops-Gerrits, A. Verberckmoes, R. Schoonheydt, M. Ichikawa and P. A. Jacobs, Microporous Mesoporous Mater., 1998, 21, 275–286 CrossRef.
- N. V. Beznis, B. M. Weckhuysen and J. H. Bitter, Catal. Lett., 2010, 136, 52–56 CrossRef CAS.
- G. B. Shul'pin, T. Sooknoi, V. B. Romakh, G. Suss-Fink and L. S. Shul'pina, Tetrahedron Lett., 2006, 47, 3071–3075 CrossRef PubMed.
- P. J. Smeets, J. S. Woertlink, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Inorg. Chem., 2010, 49, 3573–3583 CrossRef CAS PubMed.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- C. Hammond, N. Dimitratos, R. L. Jenkins, J. A. Lopez-Sanchez, S. A. Kondrat, M. H. Ab Rahim, M. M. Forde, A. Thetford, S. H. Taylor, H. Hagen, E. E. Stangland, J. H. Kang, J. M. Moulijn, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 689–699 CrossRef CAS.
- C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, M. H. Ab Rahim, M. M. Forde, A. Thetford, D. M. Murphy, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, Chem. – Eur. J., 2012, 18, 15735–15745 CrossRef CAS PubMed.
- A. R. Overweg, M. W. J. Crajé, A. M. van de Kraan, I. W. C. E. Arends, A. Ribera and R. A. Sheldon, J. Catal., 2004, 223, 262–270 CrossRef CAS PubMed.
- K. A. Dubkov, N. S. Ovanesyan, A. A. Shteinman, E. V. Starokon and G. I. Panov, J. Catal., 2002, 207, 341–352 CrossRef CAS.
- L. J. Lobree, I. C. Hwang, J. A. Reimer and A. T. Bell, J. Catal., 1999, 186, 242–253 CrossRef CAS.
- M. M. Forde, R. D. Armstrong, C. Hammond, Q. He, R. L. Jenkins, S. A. Kondrat, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. Willock, C. J. Kiely and G. J. Hutchings, J. Am. Chem. Soc., 2013, 135, 11087–11099 CrossRef CAS PubMed.
- M. M. Forde, L. Kesavan, M. I. Bin Saiman, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, S. H. Taylor, C. J. Kiely and G. J. Hutchings, ACS Nano, 2014, 8, 957–969 CrossRef CAS PubMed.
- B. Caussat and C. Vahlas, Chem. Vap. Deposition, 2007, 13, 443–445 CrossRef CAS.
- T. Tano, H. Sugimoto, N. Fujieda and S. Itoh, Eur. J. Inorg. Chem., 2012, 4099–4103 CrossRef CAS.
- A. A. Battiston, J. H. Bitter, W. M. Heijboer, F. M. F. de Groot and D. C. Koningsberger, J. Catal., 2003, 215, 279–293 CrossRef CAS.
- E.-M. El-Malki, R. A. Van Santen and W. M. H. Sachtler, J. Phys. Chem. B, 1999, 103, 4611–4622 CrossRef CAS.
- D. Chen, H. Jin, Z. Wang, L. Zhang and F. Qi, J. Phys. Chem. A, 2011, 115, 602–611 CrossRef CAS PubMed.
- I. S. Ignatyev, Y. Xie, W. D. Allen and H. F. Schaefer III, J. Chem. Phys., 1997, 107, 141–155 CrossRef CAS PubMed.
- G. Centi, S. Perathoner, F. Pino, R. Arrigo, G. Giordano, A. Katovic and V. Pedulà, Catal. Today, 2005, 110, 211–220 CrossRef CAS PubMed.
- T. Inui, H. Nagata, T. Takeguchi, S. Iwamoto, H. Matsuda and M. Inoue, J. Catal., 1993, 139, 482 CrossRef CAS.
- G. Lehmann, Z. Phys. Chem. Neue Folge, 1970, 72, 279–297 CAS.
- J. Pérez-Ramírez, J. C. Groen, A. Brückner, M. S. Kumar, U. Bentrup, M. N. Debbagh and L. A. Villaescusa, J. Catal., 2005, 232, 318–334 CrossRef PubMed.
- E. J. M. Hensen, Q. Zhu, M. M. R. M. Hendrix, A. R. Overweg, P. J. Kooyman, M. V. Sychev and R. A. van Santen, J. Catal., 2004, 221, 560–574 CrossRef PubMed.
- S. Bordiga, R. Buzzoni, F. Geobaldo, C. Lamberti, E. Giamello, A. Zecchina, G. Leofanti, G. Petrini, G. Tozzola and G. Vlaic, J. Catal., 1996, 152, 486–501 CrossRef.
- G. D. Pirngruber, P. K. Roy and R. Prins, Phys. Chem. Chem. Phys., 2006, 8, 3939–3950 RSC.
- J. Pérez-Ramírez, G. Mul, F. Kapteijn, J. A. Moulijn, A. R. Overweg, A. Domenech, A. Ribera and I. Arends, J. Catal., 2002, 207, 113–126 CrossRef.
- M. Wilke, F. Farges, P.-E. Petit, G. E. B. Jr and F. Martin, Am. Mineral., 2001, 86, 714–730 CAS.
- T. E. Westre, P. Kennepohl, J. DeWitt, B. Hedman, K. O. Hogson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6341 CrossRef CAS.
- A. A. Battiston, J. H. Bitter and D. C. Koningsberger, J. Catal., 2003, 218, 163–177 CrossRef CAS.
- J. R. Hayes and A. P. Grosvenor, J. Alloys Compd., 2012, 537, 323–331 CrossRef CAS PubMed.
- F. de Groot, G. Vanko and P. Glatzel, J. Phys.: Condens. Matter, 2009, 21, 104207–104214 CrossRef PubMed.
- J. A. Sigrist, M. W. Gaultois and A. P. Grosvenor, J. Phys. Chem. A, 2011, 115, 1908–1912 CrossRef CAS PubMed.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564 CrossRef CAS.
- H. Y. Chen and W. H. M. Sachtler, Catal. Today, 1998, 42, 73–83 CrossRef CAS.
- A. A. Battiston, J. H. Bitter, F. M. F. de Groot, A. R. Overweg, O. Stephan, J. A. van Bokhoven, P. J. Kooyman, C. van de Spek, G. Vankó and D. C. Koningsberger, J. Catal., 2003, 213, 251–271 CrossRef CAS.
- M. Kögel, R. Mönnig, W. Schwieger, A. Tissler and T. Turek, J. Catal., 1999, 182, 470–478 CrossRef.
- S. H. Choi, B. R. Wood, J. A. Ryder and A. T. Bell, J. Phys. Chem. B, 2003, 107, 11843–11851 CrossRef CAS.
- G. D. Pirngruber, K. R. Pijus and N. Weiher, J. Phys. Chem. B, 2004, 108, 13746–13754 CrossRef CAS.
- E. V. Kuznetsova, E. N. Savinov, L. A. Vostrikova and V. N. Parmon, Appl. Catal., B, 2004, 51, 165–170 CrossRef CAS PubMed.
- L. Zhao, X. Lu, Y. Li, J. Chen and W. Guo, J. Phys. Chem. A, 2012, 116, 3282–3289 CrossRef CAS PubMed.
- X. I. Sun, X. R. Huang, J. Li, R. P. Huo and C. C. Sun, J. Phys. Chem. A, 2012, 116, 1475–1485 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc00545g |
| ‡ Present address Stephenson Institute for Renewable Energy, Chemistry Department, The University of Liverpool, Crown Street, Liverpool, L69 7ZD UK. |
| § Ethanol stability over the calcined and reduced catalysts is the same. These materials show >90% ethanol decomposition to acetic acid and CO2 under simulated reaction conditions at 50 °C |
| This journal is © The Royal Society of Chemistry 2014 |