 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular control of selectivity in transition-metal catalysis through substrate preorganization
Paweł
Dydio
and
Joost N. H.
Reek
*
Homogeneous, Supramolecular and Bio-Inspired Catalysis, van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.n.h.reek@uva.nl; Fax: +31 20 5255604; Tel: +31 20 5256437
First published on 29th January 2014
Abstract
Supramolecular chemistry exploits multiple weak intermolecular interactions to assemble nano-sized molecular architectures, providing new possibilities for (transition metal) catalyst development. In this Perspective we focus on the application of such weak (directional) interactions between a substrate molecule and a (bifunctional) catalyst for structural preorganization prior to the catalytic reaction. As we discuss, such effects together with the confinement properties of the nano-space of the ‘active sites’ play a crucial role for the exceptional selectivities and activities of natural enzymes. We will elaborate on the application of such supramolecular strategy to the more traditional transition-metal catalysis, and we will compare it with the traditional substrate preorganization methods. Subsequently, literature examples of such bifunctional catalyst systems will be described in which the function of weak interactions was carefully designed a priori, as well as, the serendipitously found catalysts in which the presence of supramolecular effects was recognized post factum. The discussed examples demonstrate the power of the strategy for the control of selectivity in various types of metal catalyzed reactions, and the observation of the serendipitous findings can help to generate new leads for more efficient catalyst design.
 Paweł Dydio | Paweł Dydio (1985) studied Natural Sciences and Mathematics at the University of Warsaw, Poland (2004–2009), graduating in Chemistry (summa cum laude). While at UW, he did undergraduate research in supramolecular chemistry with Professor Janusz Jurczak. Subsequently, he joined the group of Professor Joost Reek at the University of Amsterdam, the Netherlands, and working on the discovery and development of supramolecular strategies in homogeneous transition-metal catalysis he completed his PhD (cum laude) in 2013. He is currently a postdoctoral researcher in the group of Professor John Hartwig at the University of California Berkeley, USA. |
 Joost N. H. Reek | Joost Reek (1967) completed his PhD in Nijmegen in 1996 in the area of supramolecular chemistry with Prof. Nolte. After a postdoctoral fellowship in Sydney, Australia, with Prof. M. Crossley, he joined the group of Prof. van Leeuwen at the University of Amsterdam as an assistant professor in 1998, with his research activities focusing on transition-metal catalysis. In 2005 he was elected to become a member of The Young Academy of Science (KNAW). In 2006 he became full professor (chair in supramolecular catalysis). He has received various prestigious grants, including VICI (2006) and an ERC advanced grant (2013). In 2009 he also started a spin-off company, InCatT, to explore the commercial potential of new supramolecular strategies for combinatorial approaches to find catalysts for (asymmetric) chemical transformations. In 2013 he became an elected member of Royal Holland Society of Sciences and Humanities (KHMW), and scientific director of the van 't Hoff institute for molecular sciences. His current research activities include transition-metal catalysis, supramolecular catalysis and catalysis for green energy applications. |
1. Introduction
Rapidly rising consumption of goods due to the ever increasing human population and the rise of living standards put enormous demands on the natural resources of the planet, which, if explored at the current rate, are expected to run out in the near future. This state of affairs calls for devoting more attention towards renewable resources and for using our current reserves of natural resources more efficiently. This requires the development of new technologies that are more environmentally friendly, and if possible also more economically efficient and as such attractive for industrial application. Catalysis is a key tool to control chemical transformations, especially in industry, and is therefore of crucial importance for sustainable development.1 The central role of (transition metal) catalysts is to accelerate reactions that are otherwise very slow or even impossible, primarily by creating new reactivity pathways via a combination of simple elementary steps at the metal center.2 Therefore, it can give access to new transformations and open up otherwise inaccessible, yet effective, shortcuts relevant to current synthetic schemes. In addition, on the long term new catalytic routes will be crucial for the conversion of renewable building blocks, replacing fossil-fuel-based materials.Homogeneous transition-metal catalysis offers powerful and straightforward methods for carrying out selective and effective chemical transformations,3 and as such this research field attracts considerable academic attention that has resulted in tremendous progress in the last few decades. For transition-metal catalysts the activity, selectivity (and the stability) is highly dependent on the ligands coordinated to the catalytic metal center.4 Notable insights in various reaction mechanisms including the role of the ligands, has been obtained. Computational methods become increasingly sophisticated, and yet the ab initio design of a selective catalyst for most reactions with substrates of interest is still beyond our abilities. Therefore, the search for catalysts typically still involves the knowledge-supported trial-and-error screening of candidate systems. Often, the screening of catalysts based on privileged ligands5 – those that have been proven to provide good selectivities (and activities) for a broad range of reactions and substrates – is a good starting point. If required, further structure optimization of the catalyst follows to finally obtain the catalyst with desired properties. This approach has provided many successes, but is not a general strategy that always results in success. There are reactions for which selectivity issues are difficult to solve with this approach; reactions, for example, in which the pathway to the desired product is higher in energy than the alternative pathways, or reactions with many competing pathways. Therefore, complementary approaches that allow for a more rational catalyst design for these challenging reactions would be of high value.
Supramolecular chemistry describes chemical systems formed by a self-assembly of a number of molecular building blocks or components via reversible, relatively weak interactions.6 Enormous progress in this field has been achieved over the past few decades resulting in good understanding and insight into these weak, yet effective interactions that allow for formation of large self-assembled molecular architectures.7 Therefore, supramolecular chemistry can provide powerful new tools for catalyst development and has attracted considerable attention in the last few years, giving rise to a group of strategies collectively called ‘supramolecular catalysis’.8 Much attention has been devoted to supramolecular capsules9 the interior of which mimics the finite microenvironments of natural enzymes.10 Such synthetic cavities can be used as ‘molecular flasks’ inside of which the reactions experience a series of ‘confinement effects’ that are otherwise not present in the bulk.11 This approach has resulted in a number of elegant examples of capsule-driven reactions that display enhanced selectivity and/or activity.11,12 For example, the Diels–Alder reactions within the octahedral coordination capsules, developed by Fujita and co-workers,12d,13 lead to products that are not formed otherwise in these reactions in the bulk, demonstrating the capsule-directed selectivity effect.14,15 Raymond and co-workers showed that the rate of the acid-catalyzed hydrolysis reaction of orthoesters is increased dramatically by the special microenvironment of the cage, and occurs even in a basic reaction medium,16 demonstrating the potential of the strategy for the rate enhancement. Supramolecular interactions are also widely used to construct “supramolecular bidentate ligands” from easily accessible monodentate components that are programmed to self-assemble in solution.17 The strategy gives easy access to wide libraries of ligands, particularly suitable for catalyst development based on high throughput screening and combinatorial methods. These methodologies are already well-matured, complement traditional approaches, and in principle are ready to tackle practical problems for homogeneous catalysis.18 Besides such approaches for which the supramolecular tools are used just to construct the catalysts from simpler subcomponents by self-assembly, recent breakthroughs demonstrate that application of supramolecular interactions between a catalyst and a substrate can be extremely powerful. Considering that the selectivity of catalytic reactions is determined by energy differences in the competing transition states that are as small as 3 kcal mol−1, one can envision that the relatively weak supramolecular interactions can be used to change the selectivity of any catalytic transformation. In such approach the aim is no longer to affect the catalyzed reaction by just modifying secondary coordination sphere of the catalyst, but supramolecular chemistry tools are applied to control directly the chemical transformation taking place at the metal center via precise positioning of the substrate molecule. In this Perspective we will highlight some of the recent examples to demonstrate the power of this strategy.
2. Confinement effects and active substrate preorganization in natural enzymes
Nature has served as a major source of inspiration in the area of supramolecular chemistry. Correspondingly, enzymes – natural catalysts – have served as models for the design of supramolecular catalysts. In enzymes, an ‘active site’ at which the catalytic reaction takes place is typically buried within a specific proteomic microenvironment. Therefore, a substrate that is brought into a confined space of this ‘cavity’ experiences a series of ‘confinement effects’ that are otherwise not present in the bulk.10 For instance, the encapsulated substrate molecule can accept only specific conformations imposed by the size and shape of the cavity that also limit its motion, and restricting the number of possible reactions. This usually also results in reduced activation entropy of a reaction. The proximity and orientation of the reactive groups is usually restricted, which in turn determines the reaction selectivity. In some cases, the substrate or the reaction intermediate is forced to adopt a high-energy conformation required for increased reactivity,19 which can effectively lower the free-energy reaction barrier, accelerating the reaction. Besides, the transition state of the effective reaction pathway can be stabilized more efficiently than the substrate in its ground state by the surrounding cavity, decreasing the overall energy barrier of the reaction. Mimicry of such effects was intensively explored with synthetic supramolecular capsules. This brought a number of elegant capsular catalysts that display unusual capsule-driven selectivity and/or enhanced activity in the catalyzed reactions.11,12 In natural enzymes, however, aside from the confinement effects intrinsic to the nano-space of the active sites, other factors often play an important role in controlling the selectivity and the activity of the catalyzed reaction. Frequently the positioning of the substrate is determined not only by the size and shape of the enzymatic cavity, but also by directional interactions between functional groups of the substrate and of the active site, such as hydrogen bonding, ionic interactions or even reversible covalent bonding.10 This ensures precise substrate orientation and dramatically limits its motion, both of which are essential for obtaining high selectivity. Importantly, this operational mode is less susceptible to changes in substrate size (as long as a substrate molecule bears the essential functional groups), and allows for selective transformations of a broad substrate scope. In contrast, substrate orientation based exclusively on shape and size should in principle be more substrate dependent, as is seen, for instance, for the Diels–Alder reactions within the cage-type catalysts.14,15 Furthermore, such interactions with the functional groups of the substrate can substantially modulate their reactivity allowing for even better control over the selectivity and the activity of the reaction. Importantly, these directional interactions are well-understood and hence can be precisely manipulated, allowing for the rational design of the ‘active sites’ of such synthetic mimics of enzymes.3. Traditional vs. bio-inspired supramolecular substrate preorganization
In traditional catalysis, substrate preorganization is realized via coordination of a ‘directing group’ to a metal center (Scheme 1).20 The directing group can be either a functional group present in the substrate or it is temporarily introduced before the catalytic reaction. Although effective, this methodology is limited to substrates with directing groups spatially close to the reactive functionality, imposing limitations. Moreover, the reaction occurring at the metal center must be compatible with the directing group, and also requires one vacant coordination site at the metal center, which cannot be realized for all reactions.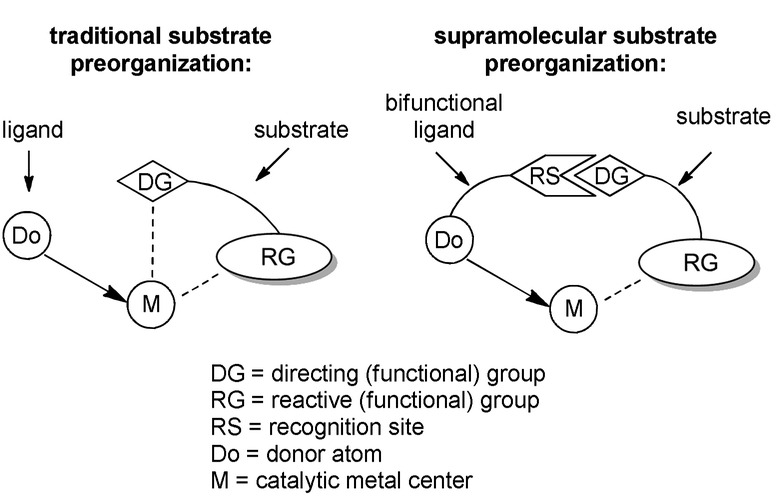 | ||
| Scheme 1 Schematic representation of traditional and supramolecular substrate preorganization. | ||
Supramolecular chemistry provides ideal tools for substrate preorganization. In transition-metal catalysis, substrate preorganization can be realized by the supramolecular interactions between the ‘directing group’ of the substrate and a suitable bifunctional ligand from the catalyst (Scheme 1). In principle, a catalyst that is furnished with a specific recognition site will bind the substrate molecule and preorganize its reactive moiety with respect to the metal center. The location of the directing group and the reactive functionality are theoretically independent. This makes it possible for the strategy to be applied to substrates with directing groups in various positions of the molecule, allowing for remote control, and exceeding the possibilities of the traditional substrate preorganization approach. Thanks to a well-established understanding of weak interactions between molecules, the rational design of selective catalysts that operate predictively can be envisioned. The methodology is especially attractive for reactions for which the selectivity issues are notoriously difficult to solve with the traditional trial-and-error approach, e.g. reactions for which the pathway to the desired isomer is higher in energy than the alternative one or for which many reaction pathways compete simultaneously.
3.1 Non-covalent substrate preorganization
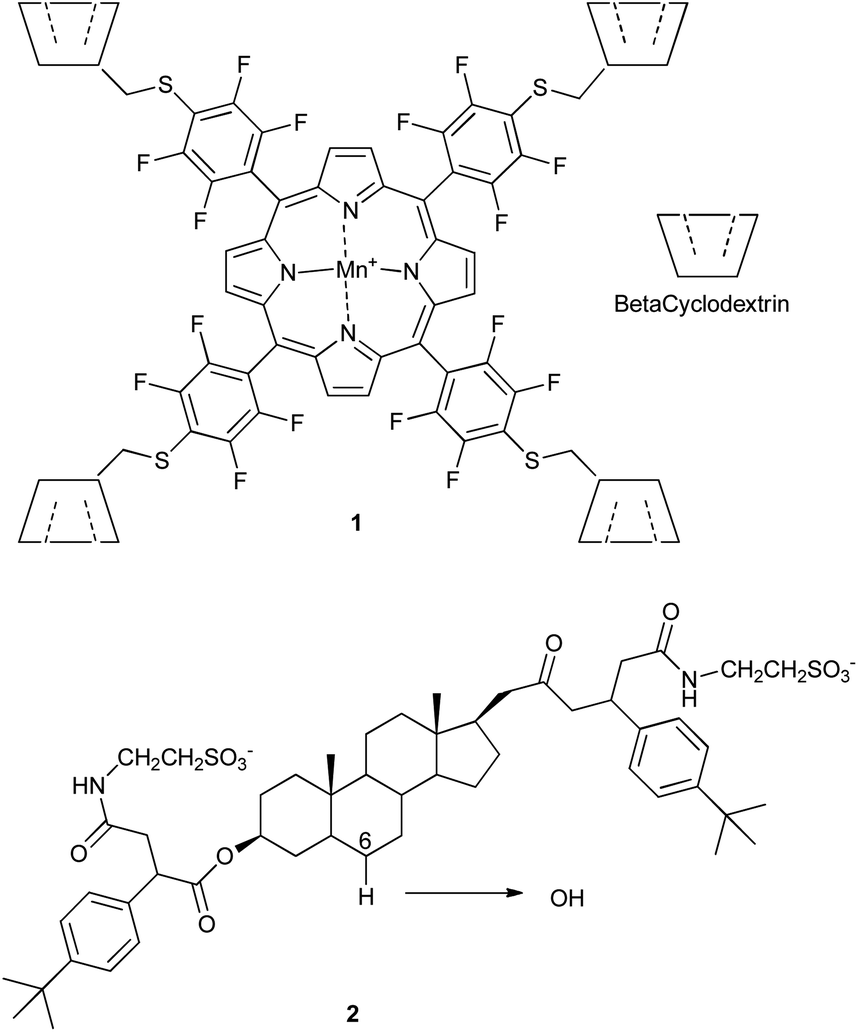 | ||
| Scheme 2 Mn-catalyst 1 with attached cyclodextrins, and selective hydroxylation of steroid 2 catalyzed by 1. | ||
In another study, another Mn-catalyzed, selective C–H oxidation at sp3-carbon atoms was targeted. In this case nonporphyrin system 3 was devised, which consists of a di-μ-oxo dimanganese core – a very active catalyst for oxidation chemistry – based on ligand 4 equipped with Kemp's triacid, a recognition site for carboxylic acids (Fig. 1).23 Molecular modeling predicts that the catalyst binds a molecule of ibuprofen (5) in such a way that the benzylic proton of the more remote position is precisely oriented towards the catalytic manganese center (Fig. 1). As predicted by the model, the catalyst oxidizes ibuprofen at this position with very high regioselectivity (98.5%, Scheme 3). In contrast, the reaction with a catalyst devoid of the COOH recognition site leads to a mixture of products. Impressively, catalyst 3 can also oxidize (4-methylcyclohexyl)acetic acid 6 selectively (>99%) at the C(4)–H position, as predicted by the modeling. Interestingly, only the trans isomer of 6 reacts readily, since the cis isomer does not expose an oxidisable C–H group to the catalytic center when its COOH is bound to the recognition site. In a control experiment with the catalyst devoid of the COOH recognition site both isomers react similarly, and the reaction leads to a mixture of several oxidation products. This system shows that, along with providing an unprecedented level of reaction selectivity, bifunctional catalysts can allow for efficient substrate selection, including challenging diastereoselection.
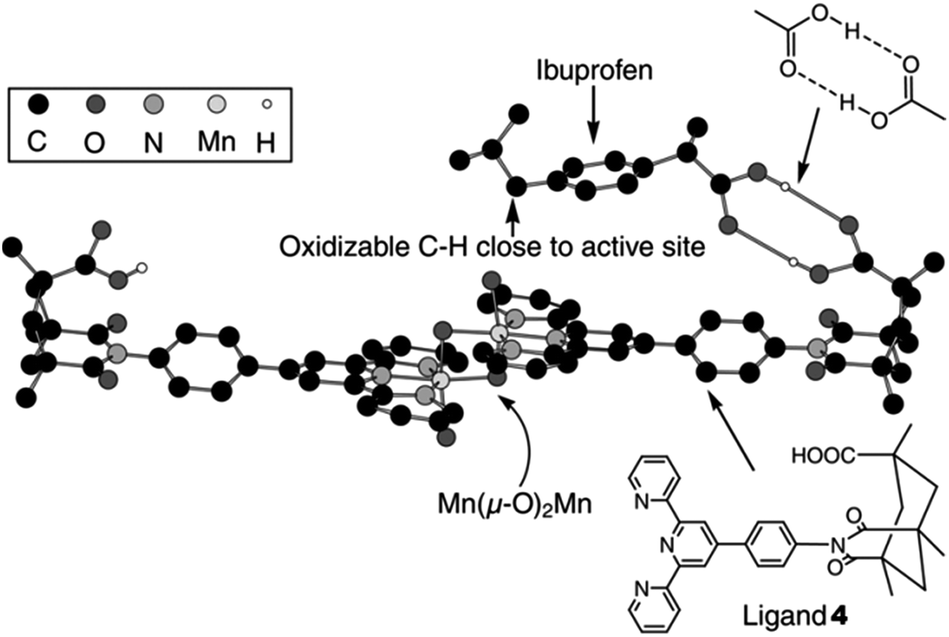 | ||
| Fig. 1 Molecular model of catalyst 3 docked with a molecule of hydrogen-bonded substrate 5 – Ibuprofen. Reprinted with permission from ref. 23. Copyright 2006, American Association for the Advancement of Science. | ||
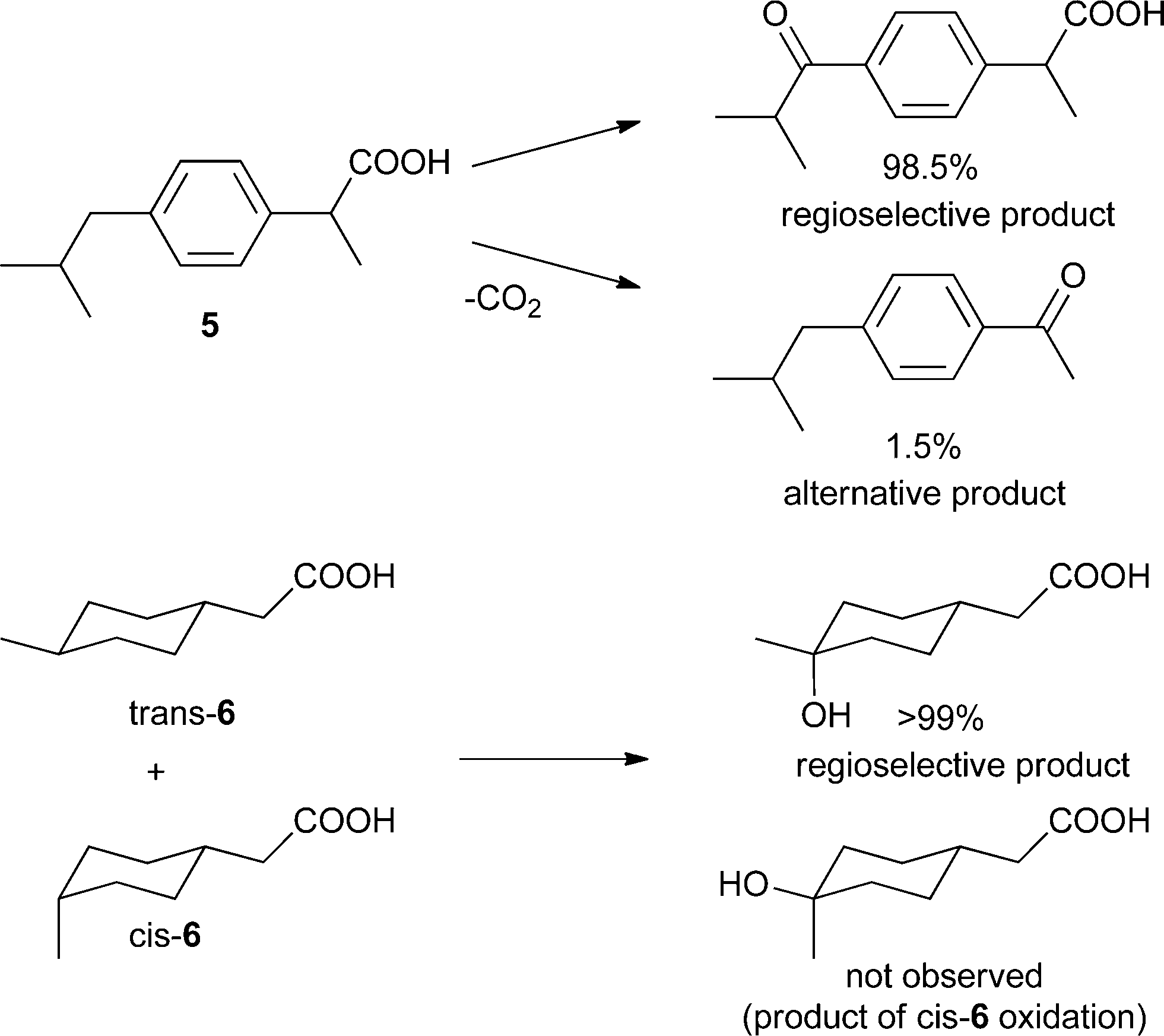 | ||
| Scheme 3 Oxidation products of substrates 5–6 with 3 as catalyst. | ||
In a subsequent study, the same catalyst and an analogue – a manganese porphyrin catalyst furnished with the same Kemp's triacid recognition site – were used as catalysts for selective epoxidation of alkenes containing a carboxylic group.24 For two out of three substrates studied, molecular recognition directs oxidation to the olefin moiety and prevents the unselective oxidation of C–H bonds observed in control experiments. However, poor diastereoselectivity is observed for the epoxide products, which on the basis of molecular modeling is attributed to the ill-defined preference between alternative orientations of the reactive double bond in the catalyst–substrate complex.
In a related study, a Ru(II)–porphyrin 7 equipped with a chiral tricyclic γ-lactam that can form a complementary two-point hydrogen bonding with the CONH amide functional group was synthesized (Fig. 2).25 Molecular modeling showed that this two-point interaction orients 3-vinylquinolone (8) at the catalyst (Fig. 2) such that one prochiral site of the reactive double bond is exposed to the catalytic center preferentially due to steric requirements. As predicted by the model, substrate preorganization leads to high enantioselectivities (ee's up to 98%) in epoxidation for a series of 3-vinylquinolones. The epoxidation of substrate 9 with two reactive sites, 3,9-divinylquinolone, takes places with high regioselectivity on the vinyl group in position 3 (91% vs. 9% of the alternative product, Scheme 4). This selectivity is in line with the proposed mechanistic model, which predicts that only the vinyl group in position 3 can react at the catalytic center when the substrate is bound to the recognition site. The reaction with the same substrate, but with a catalyst devoid of the recognition site, leads to the two alternative products in a 62/38 ratio, revealing the crucial role of substrate orientation for the selectivity. The importance of the double hydrogen bonding was further confirmed by control reactions with either the N-methylated substrate or catalyst, which leads in both experiments to drastic deterioration of the selectivity. Studies on the substrate scope show that other substrates, such vinylpyridones, primary alkenoic amides and carbamates also experience the effect of the preorganization. However, a clear trend can be noted whereby the enantioselectivity gradually drops with increasing flexibility of the substrate. The distance between the catalytic metal and the chiral groups is over 7.0 Å, showing that the remote selectivity control via supramolecular interactions is viable.
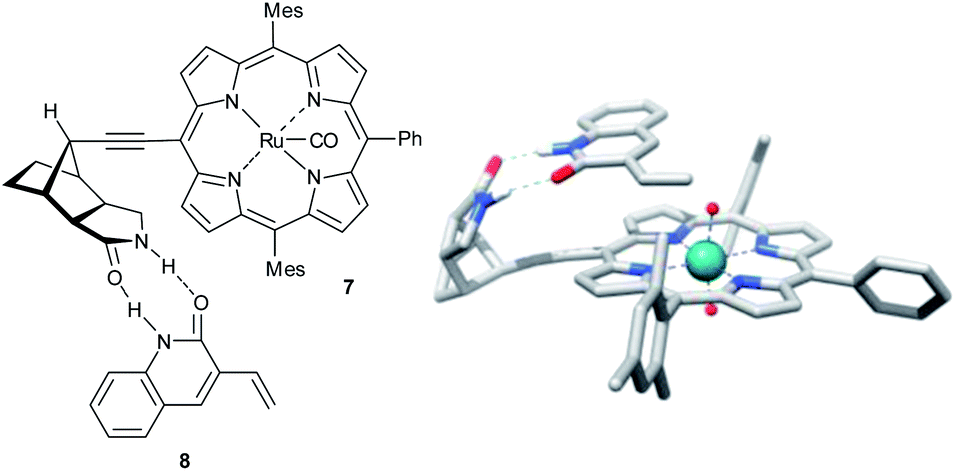 | ||
| Fig. 2 Ru(II)–porphyrin pre-catalyst 7 docked with substrate 8 – schematic representation and the probable transition state found by DFT. | ||
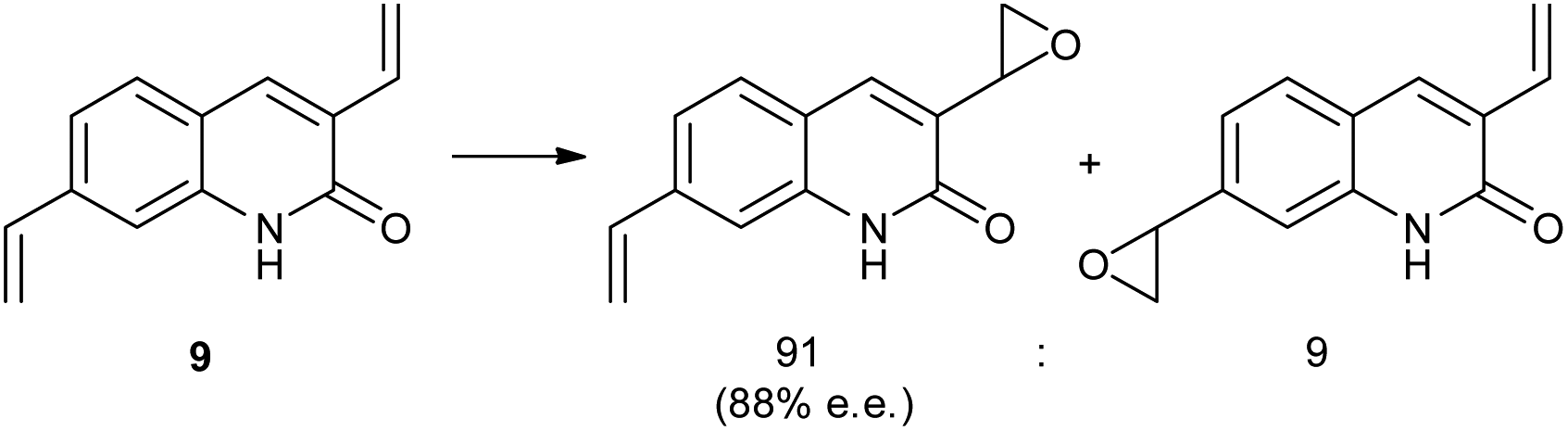 | ||
| Scheme 4 Epoxidation products of substrate 9 with 7 as catalyst. | ||
In a subsequent study, the same recognition site was used to preorganize sulfide quinolone-type derivatives on a Mn(III)–salen catalyst, resulting in an enantioselective sulfoxidation reaction (ee's up to 71%).26 Again, control experiments confirmed the crucial role of the two-point hydrogen bonding in the prochiral face differentiation.
Recently it was also reported that a Rh(II) complex bearing the same two γ-lactam binding sites catalyzes the regio- and enantioselective (ee's up to 74%) C–H amination reactions of 3-benzylquinolones.27 It was confirmed that both the regio- and enantioselectivity of the reaction is controlled by substrate positioning imposed by the hydrogen bonding.
The chiral tricyclic γ-lactam binding site was also used to preorganize prochiral substrates at photosensitizers (in metal-free reactions). This enabled unprecedented induction of enantioselectivity in the photoinduced conjugate additions of α-amino alkyl radicals to enones,28 as well as in intra- and intermolecular [2 + 2] photocycloaddition reactions.29,30
In another study, regioselectivity issues in hydroformylation reactions were addressed with phosphine ligand 10 bearing a guanidine-based recognition unit that is able to bind unsaturated carboxylic acids via two-point hydrogen bonding (Scheme 5).31 The substrate binding results in increased activity and high regioselectivity for hydroformylation of 3-alkenoic acids 11–13. Remarkably, the system displays unprecedented high regioselectivity for hydroformylation of an internal alkene 12, introducing the aldehyde group with high selectivity at the carbon atom of the double bond more distant from the carboxylic moiety. In addition, the catalyst can discriminate between two similar reactive double bonds of substrate 13 by substrate preorganization (Scheme 5). The crucial role of binding to the recognition unit of the ligand was demonstrated by a series of control experiments with substrates or catalyst derivatives with modified ligands that cannot form the supramolecular interaction. The system is highly susceptible to the relative position of the directing carboxylic group and the double bound, as the hydroformylation of an analogue of 11 that is one carbon longer (4-pentenoic acid 14) leads to a typical mixture of isomeric products. 2-Alkenoic acid 15 undergoes 2-regioselective decarboxylative hydroformylation, resulting in formation of the linear aldehyde, thus formally substituting the carboxylic group with an aldehyde (Scheme 5).32 Furthermore, a related guanidinium-based system was used for aldehyde hydrogenation and for tandem hydroformylation–hydrogenation of terminal olefins, again with a crucial role for the hydrogen bonding functional groups.33
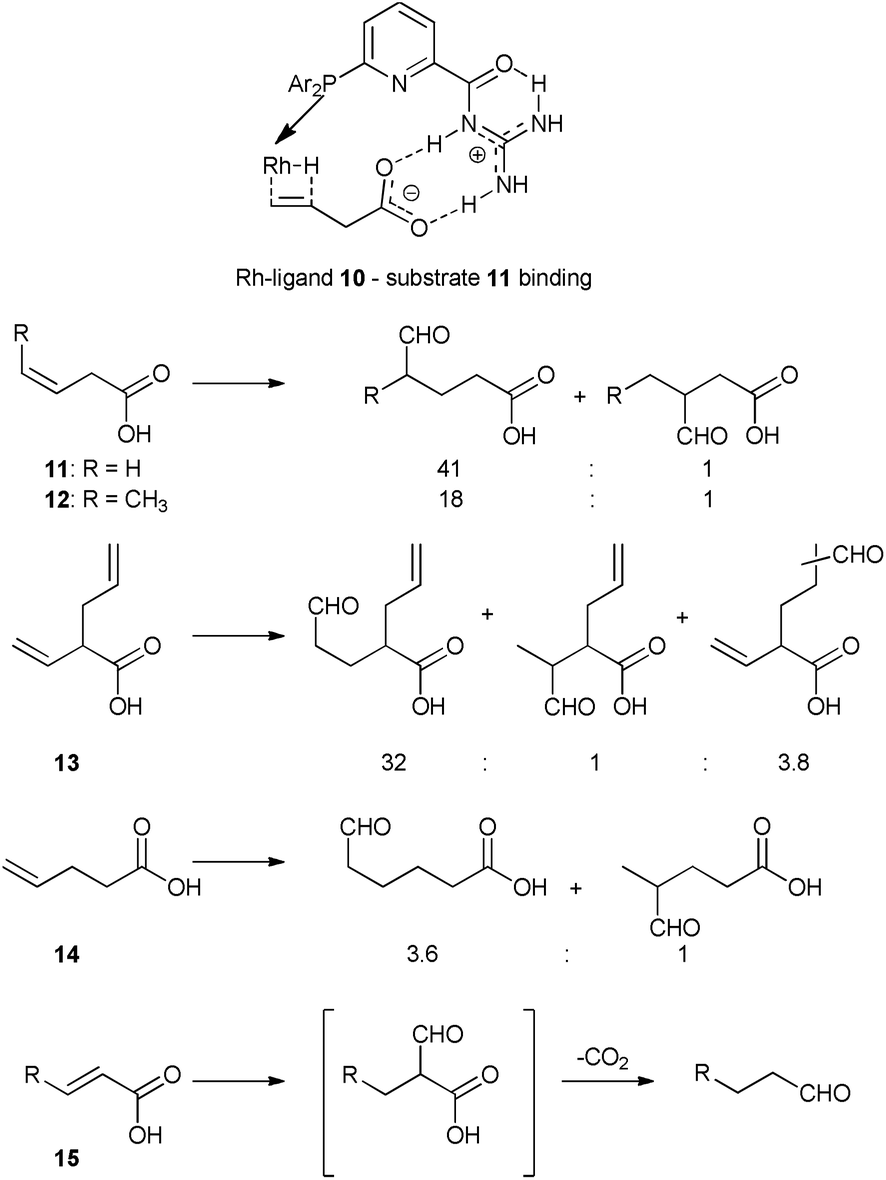 | ||
| Scheme 5 Binding of substrate 11 to ligand 10 in the hydride migration step of hydroformylation (above), and hydroformylation products of substrates 11–15 with Rh–10 catalyst (below). | ||
Highly regioselective hydroformylation of unsaturated acids was also achieved with a series of bidentate phosphorus ligands – DIMPhos ligands – that are equipped with an integral binding site for anions such as carboxylates (the DIM pocket).34 It was shown that under hydroformylation conditions the ligands bind to a rhodium center in a bidentate fashion forming the typical rhodium–hydride complexes that are active in hydroformylation. Importantly, the metal complexes can strongly bind the anionic species in the DIM binding site of the ligand, without affecting the metal coordination sphere. The catalytic studies demonstrate that substrate preorganization results in unprecedented selectivities in hydroformylation of a broad range of terminal and internal alkenes functionalized with an anionic carboxylate or phosphate group (Scheme 6). Remarkably, the selectivity controlling anionic group can be even 10 bonds away from the reactive double bond, demonstrating the power of the supramolecular substrate orientation approach. Control experiments, in which formation of the hydrogen bonding is hindered, confirm the crucial role of the substrate binding to the DIM pocket for the selectivity. DFT studies reveal that the substrate binding in the DIM pocket restricts the rotational freedom of the reactive double bound (Fig. 3). As a consequence, the pathway to one product is strongly hindered, whereas that for the desired product is lowered in energy. Detailed kinetic studies, together with the in situ spectroscopy and the isotope-labelling studies support this mechanistic model and reveal that the supramolecular system follows enzymatic-type Michaelis–Menten kinetics, with competitive product inhibition. The system was further combined with an isomerization catalyst in the one-pot tandem isomerization–hydroformylation of terminal olefins leading to valuable α-methyl-branched aldehyde products with unprecedented selectivity.35
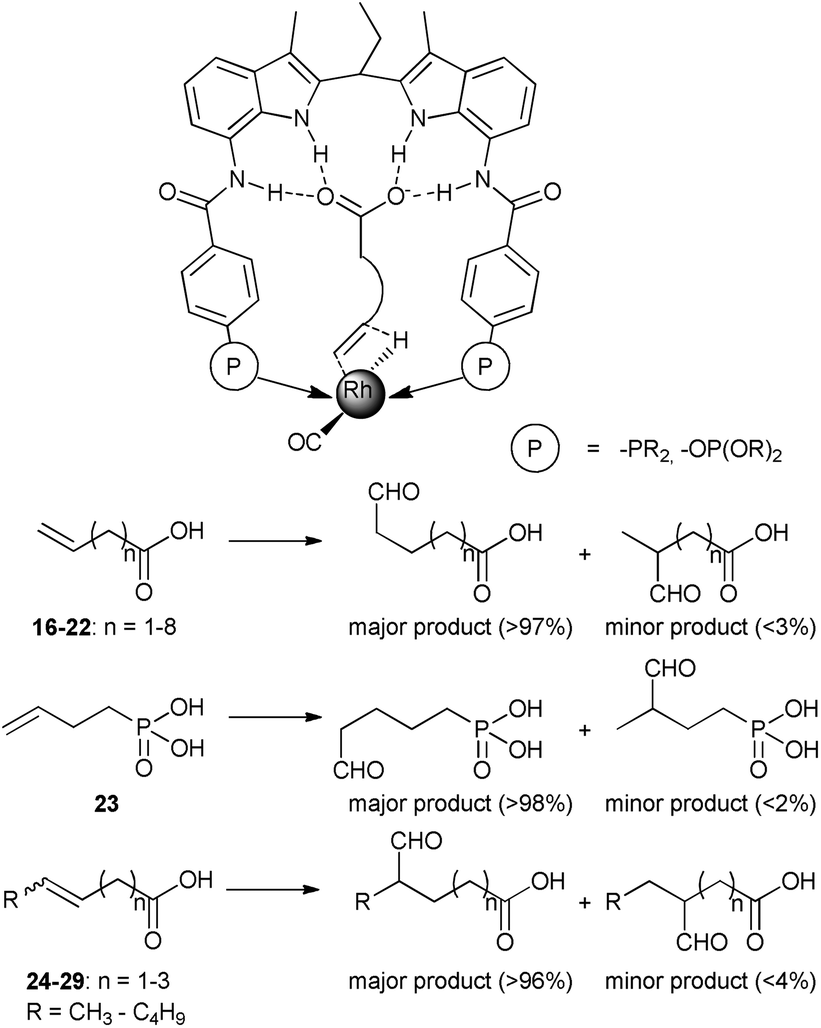 | ||
| Scheme 6 Binding of substrates to Rh–DIMPhos catalyst in the hydride migration step of hydroformylation (above), and hydroformylation products of substrates 16–29 with Rh–DIMPhos catalyst (below). | ||
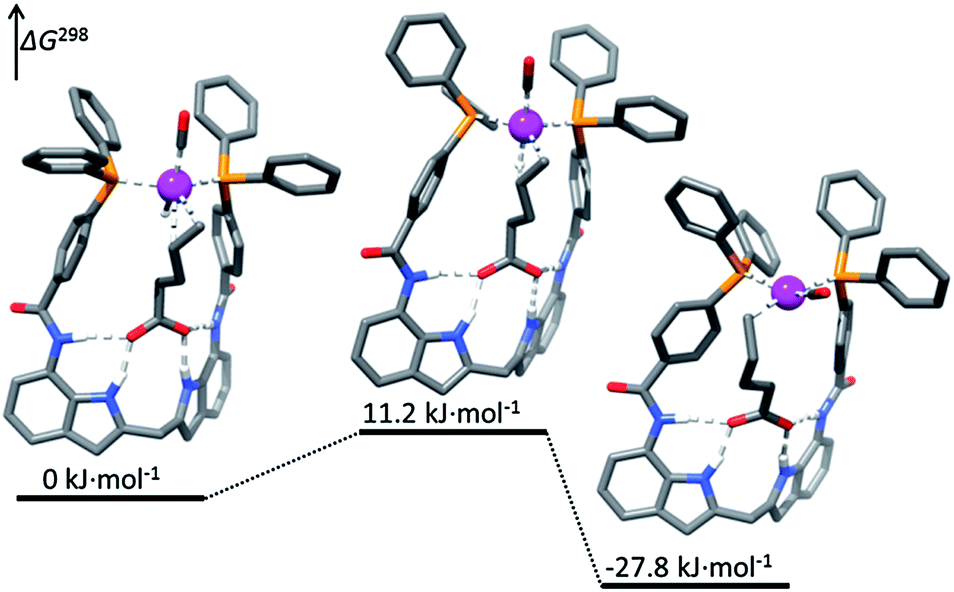 | ||
| Fig. 3 Substrate orientation during the regioselectivity-determining hydride migration step in the hydroformylation of substrate 17 with Rh–DIMPhos catalyst (DFT studies). Reprinted with permission from ref. 34b. Copyright 2013, American Chemical Society. | ||
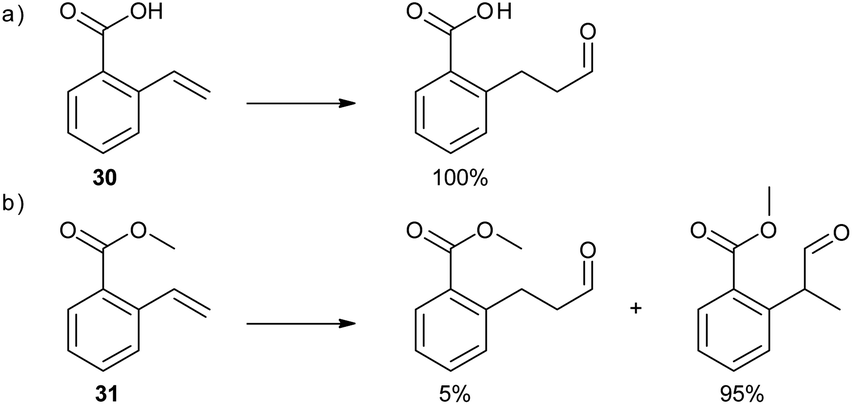
In a subsequent study, the DIMPhos catalyst system was used to control the regioselectivity in the hydroformylation of vinyl aryl derivatives.36 Common hydroformylation catalysts generally produce the α-aldehyde products, due to the electronic properties of this class of substrate, and catalysts that overrule this natural selectivity to form preferably the linear aldehyde are scarce.37 Remarkably, supramolecular substrate preorganization in the DIM pocket of the catalyst fully reverts the regioselectivity, and the β-aldehyde isomer is formed exclusively for substrate 30 (Scheme 7a). In sharp contrast, if the hydrogen bonding interaction is disrupted, for example in case of O-methylated substrate 31, the α-aldehyde is formed as the notably major product (95%), along with only a minor quantity of the β-isomer (Scheme 7b). Of note, substrate 30 reacts several times faster than ester analogue 31, demonstrating the effect of substrate preorganization on the rate of the reaction. The DIMPhos catalyst proved to be selective for a wide scope of vinyl arene substrates, including the most challenging substrates with an internal double bond. Importantly, this unprecedented selectivity opens new synthetic routes to valuable common intermediates for important synthetic targets, showing the added value of the supramolecular chemistry to the practical synthesis of fine-chemicals.
 | ||
| Scheme 7 Influence of substrate preorganization: (a) hydroformylation of substrate 30 that binds to the DIMPhos ligand, and (b) hydroformylation of substrate 31 that cannot bind to the DIMPhos ligand (both with Rh–DIMPhos as catalyst). | ||
Inspired by the cooperativity displayed by metalloenzymes such as carboxypeptidase, capable of peptide bond hydrolysis, highly active and selective bifunctional catalysts 32 for anti-Markovnikov hydration of terminal alkynes were developed (Scheme 8).38 The full reaction mechanism has not yet been revealed, however, studies so far have shown the crucial role of pendant basic functional groups that take part in proton transfer and hydrogen bonding in the reaction intermediate.39
 | ||
| Scheme 8 Anti-Markovnikov hydration of terminal alkynes with bifunctional catalyst 32. | ||
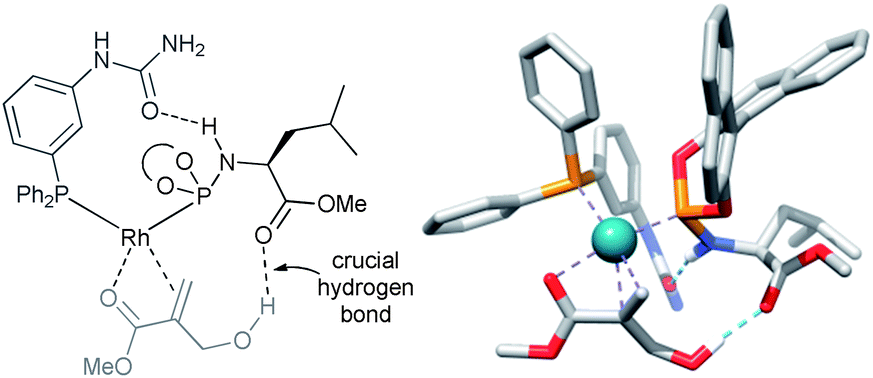 | ||
| Fig. 4 Substrate orientation through additional hydrogen bonding between the hydroxyl group of the substrate and the ester unit of the LEUPhos – determined by the DFT and control experiments. | ||
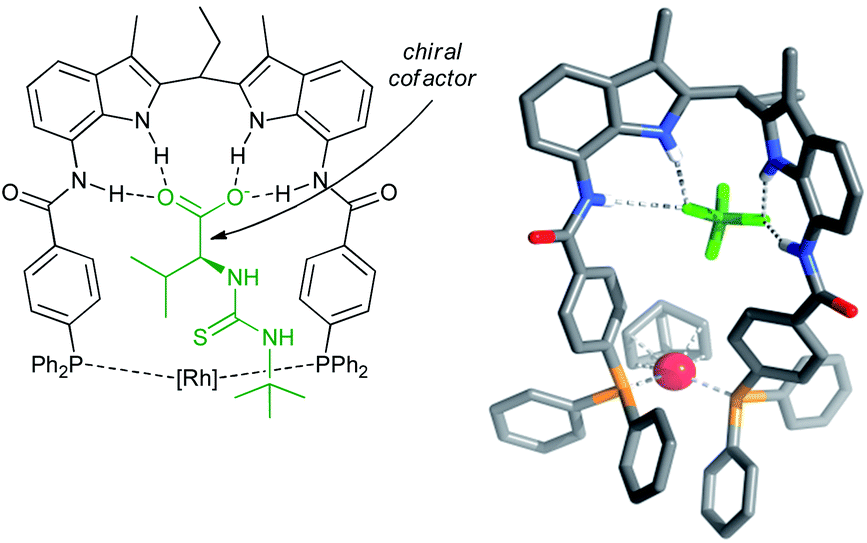 | ||
| Fig. 5 Structure of the rhodium catalyst of the ligand with the cofactor bound in the binding site (left), and crystal structure of the precatalyst (right). Reprinted with permission from ref. 42. Copyright 2011, American Chemical Society. | ||
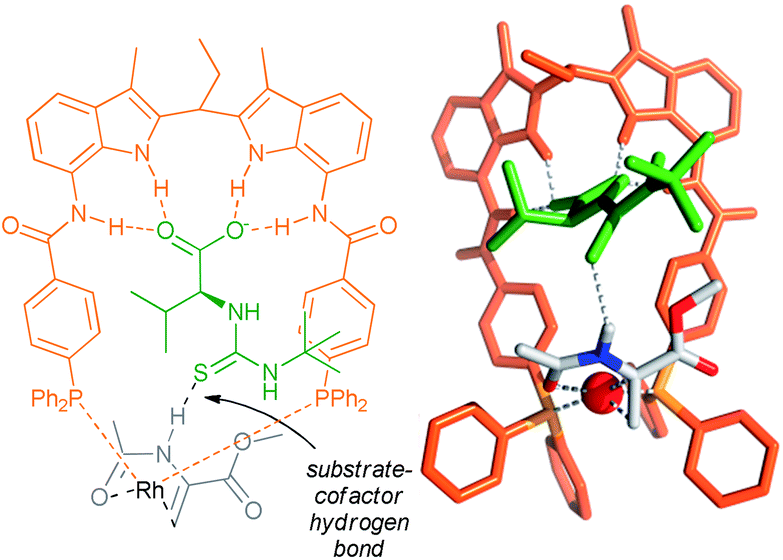 | ||
| Fig. 6 Substrate orientation through additional hydrogen bonding between the amide NH group of the substrate and the thiocarbonyl unit of the cofactor – determined by the DFT and control experiments. | ||
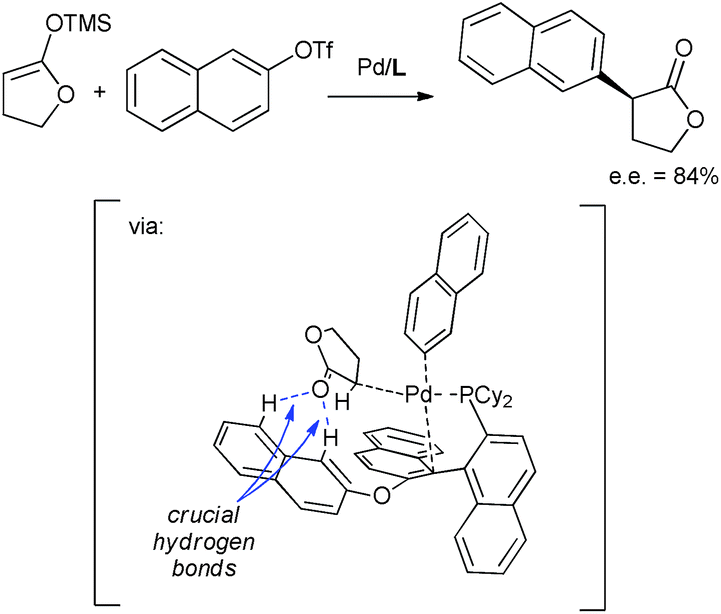 | ||
| Scheme 9 C–H⋯O hydrogen bonding mediated selectivity in palladium-catalyzed arylation of lactones. | ||
In the search for a selective (and active) catalyst for challenging allylic alkylation of indoles with unsymmetrical 1,3-disubstituted allyl acetates, P,S-bidentate ligands with an additional sulfinyl group were explored.47 It was found that the additional S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety forms a hydrogen bond with the indole–NH, and presumably helps to orient the nucleophile during the reaction (Scheme 10), resulting in a high level of regio- and enantioselectivity (ee's 84–95%). This interaction seems to be crucial not only for the selectivity but also for the activity, as the reaction with N-substituted indole, which cannot form this interaction, does not take place. This successful example of a cooperative catalyst should encourage application of bifunctional ligands in catalyst discovery.
O moiety forms a hydrogen bond with the indole–NH, and presumably helps to orient the nucleophile during the reaction (Scheme 10), resulting in a high level of regio- and enantioselectivity (ee's 84–95%). This interaction seems to be crucial not only for the selectivity but also for the activity, as the reaction with N-substituted indole, which cannot form this interaction, does not take place. This successful example of a cooperative catalyst should encourage application of bifunctional ligands in catalyst discovery.
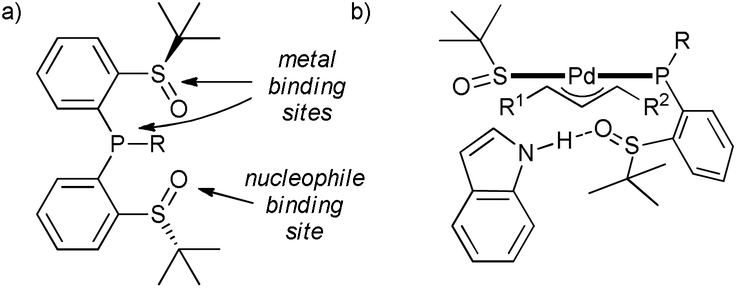 | ||
| Scheme 10 (a) Structure of the bifunctional ligands, and (b) tentative model of hydrogen-bond induced indole activation toward allylation. | ||
3.2 Covalent yet reversible substrate preorganization
The examples presented so far have primarily shown supramolecular substrate preorganization accomplished mainly through hydrogen bonding. However, in principle, any reversible bonding that does not interfere with either the metal center or the catalytic reaction can be used for that purpose. Reversible covalent bonding is an attractive tool to control selectivity of a reaction by modifying substrates with covalently attached ligand-type directing groups (e.g. diphenylphosphinobenzoate group).20a Reversible bonding allows for the use of only catalytic amounts of these kinds of directing groups, improving the applicable value of the approach (from an atom-efficient and a commercial point of view).To illustrate the potential of reversible covalent bonding, catalytic scaffolding ligands have been introduced and extensively studied in the Rh-catalyzed hydroformylation of unsaturated alcohols and sulfonamides (Scheme 11a and b).48 In principle, under optimized conditions the scaffolding ligands extensively exchange with hydroxyl functionalities of substrates. Under catalytic conditions, the formed ligand-substrate composite reacts preferentially over the free substrate, since the ligand donor moiety coordinates to the metal bringing the reactive double bond near the catalytic center. After the transformation, the product formed is exchanged for a new substrate, which subsequently reacts. This efficient substrate preorganization allows for highly regio-, diastereo- and enantioselective reactions of various terminal and internal alkenes bearing either hydroxy or sulfonamide groups.48 The strategy also facilitates hydroformylation of challenging tertiary carbon atoms that are difficult to convert otherwise, highlighting the power of the approach. Of note, the concept was previously developed to facilitate ortho-selective C–H activation and functionalization of phenol and aniline derivatives49 and hydrogenation of acrylic acid derivatives (Scheme 11c and d).50 The later study shows that scaffolding ligands allow for unprecedented (yet moderate) initial selectivity for a reaction at the C(2)C(3) double bond in hydrogenation of 2,4-hexadienoic acid, while a control reaction without the scaffolding ligand shows the initial preference for the reaction at the C(4)C(5) double bond (Scheme 11e).
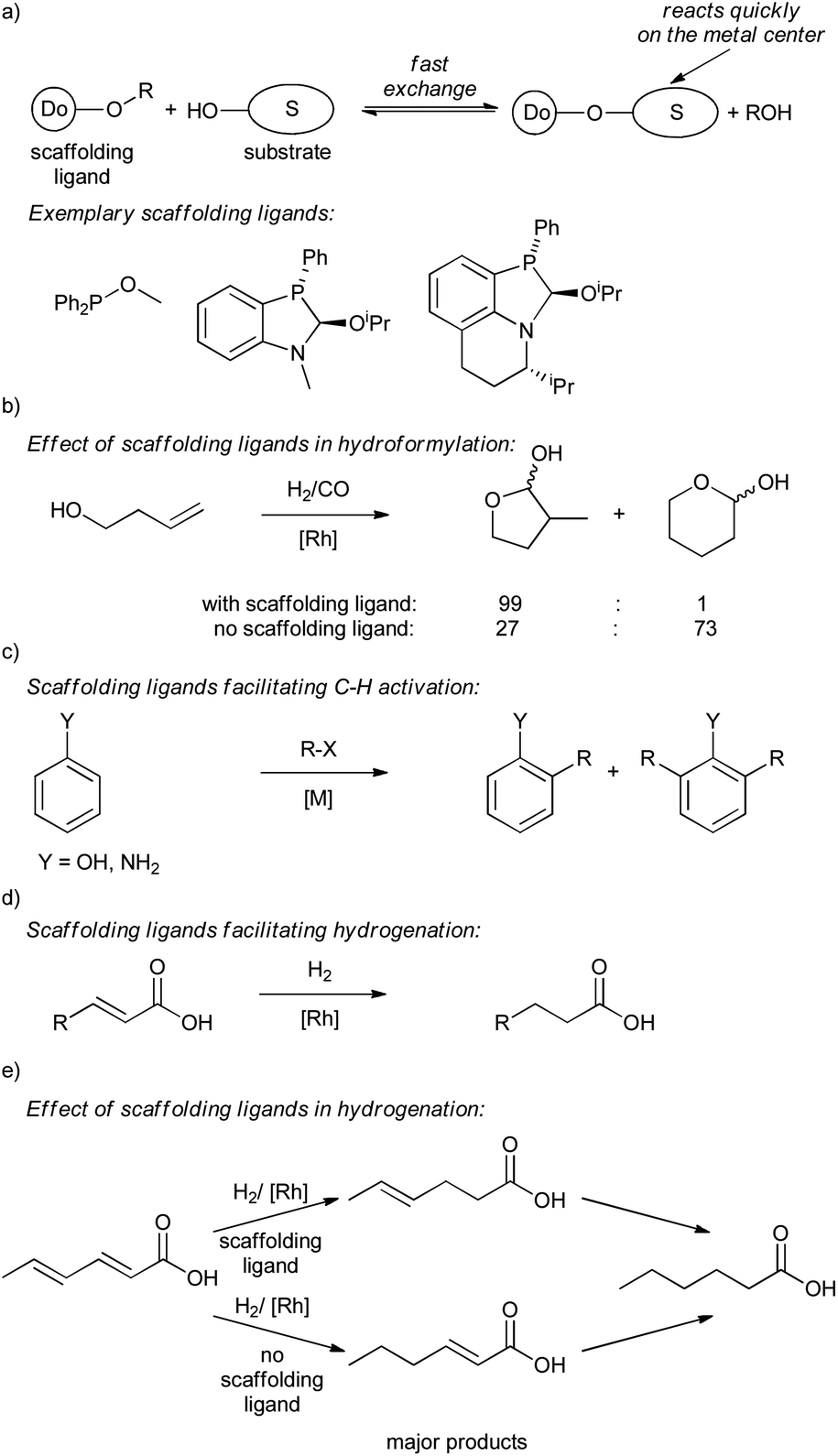 | ||
| Scheme 11 (a) Equilibrium with scaffolding ligands, (b) the effect of scaffolding ligands on reaction selectivity in hydroformylation of homoallylic alcohols, (c) scaffolding ligands facilitating C–H activation of phenols and anilines, (d) hydrogenation of 2-alkenoic acids, and (e) the effect of scaffolding ligands on reaction selectivity in hydrogenation of 2,4-hexadienoic acid. | ||
4. Conclusions and outlook
In recent years the field of supramolecular catalysis has grown significantly, providing new tools for selective transformations. Whereas initial focus was mainly on the mimicry of enzymes, generally also exploring reactions that are important in nature such as hydrolysis, currently the attention has shifted to the exploration of supramolecular tools in transition-metal catalysis. The concept of substrate organization, which is indeed also important for the operational mode of enzymes, has been broadly explored for transition-metal catalysis, and is the main topic of this Perspective. Supramolecular substrate preorganization was demonstrated to be key in a number of highly selective catalytic transformations. Importantly, for several examples, such supramolecular bifunctional catalysts provide unprecedented reaction selectivities, with reaction rates that are sufficiently high for practical applications. Hence, supramolecular substrate preorganization with bifunctionalized catalysts clearly outlines a strategy that may quickly lead to applications in industrial synthesis. As in principle, substrate orientation at the catalyst is predictable, the design of selective catalysts for specific substrates of interest is at hand. However, further mechanistic studies are required to fully understand the principles that rule the reactions carried out with such supramolecular catalysts. This will further help researchers to take full advantage of the concept and will facilitate the step to practical applications. Such studies will substantially strengthen the potential of the approach, and we foresee a bright future for supramolecular transition-metal catalysis in which substrate orientation directs the selectivity in these transformations.Acknowledgements
We kindly acknowledge NWO and NRSC-C for financial support and Dr. W. Dzik, Dr. J. I. van der Vlugt and Prof. dr. B. de Bruin for fruitful discussion.References
- M. Beller, Chem. Soc. Rev., 2011, 40, 4891–4892 RSC.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2009 Search PubMed.
- (a) Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Hand-book in Three Volumes, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, edn 2, 2002 Search PubMed; (b) Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2004 Search PubMed.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
- J. M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley-VCH, Weinheim, Germany, 1995 Search PubMed.
- Supramolecular Chemistry: from Molecules to Nanomaterials, 8 volume set, ed. J. W. Steed and P. A. Gale, Wiley-Blackwell, London, GB, 2012 Search PubMed.
- Supramolecular Catalysis, ed. P. W. N. M. van Leeuwen, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- (a) M. M. Conn and J. Rebek, Jr, Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS PubMed; (b) J. Rebek, Jr, Chem. Soc. Rev., 1996, 25, 255–264 RSC; (c) F. Hof, S. L. Craig, C. Nuckolls and J. Rebek, Jr, Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS; (d) A. Lutzen, Angew. Chem., Int. Ed., 2005, 44, 1000–1002 CrossRef PubMed; (e) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; (f) D. Ajami and J. Rebek, Jr, Acc. Chem. Res., 2013, 46, 990–999 CrossRef CAS PubMed; (g) J. Rebek, Jr, Angew. Chem., Int. Ed., 2005, 44, 2068–2078 CrossRef PubMed; (h) M. Yoshizawa and M. Fujita, Bull. Chem. Soc. Jpn., 2010, 83, 609–618 CrossRef CAS; (i) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (j) Z. Laughrey and B. C. Gibb, Chem. Soc. Rev., 2011, 40, 363–386 RSC.
- D. Ringe and G. A. Petsko, Science, 2008, 320, 1428–1429 CrossRef CAS PubMed.
- T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC.
- (a) Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC; (b) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed; (c) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed; (d) B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51–56 RSC; (e) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349–358 CrossRef CAS PubMed; (f) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (g) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef CAS PubMed; (h) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–360 CrossRef CAS PubMed; (i) A. W. Kleij and J. N. H. Reek, Chem.–Eur. J., 2006, 12, 4218–4227 CAS; (j) D. M. Vriezema, M. C. Aragones, J. A. A. W. Elemans, J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed; (k) B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, Chem.–Eur. J., 2012, 18, 12178–12190 CrossRef CAS PubMed; (l) R. Gramage-Doria and J. N. H. Reek, ChemCatChem, 2013, 5, 677–679 CrossRef CAS.
- (a) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed; (b) S.-Y. Yu, T. Kusukawa, K. Biradha and M. Fujita, J. Am. Chem. Soc., 2000, 122, 2665–2666 CrossRef CAS; (c) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- (a) T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2866–2867 CrossRef CAS PubMed; (b) S. Horiuchi, T. Murase and M. Fujita, Chem.–Asian J., 2011, 6, 1839–1847 CrossRef CAS PubMed.
- (a) M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CAS; (b) M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 11423–11429 CrossRef CAS PubMed.
- For reviews, see: (a) R. Bellini, J. I. van der Vlugt and J. N. H. Reek, Isr. J. Chem., 2012, 52, 613–629 CrossRef CAS; (b) S. Carboni, C. Gennari, L. Pignataro and U. Piarulli, Dalton Trans., 2011, 40, 4355–4373 RSC; (c) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed; (d) P. E. Goudriaan, P. W. N. M. van Leeuwen, M.-N. Birkholz and J. N. H. Reek, Eur. J. Inorg. Chem., 2008, 2939–2958 CrossRef CAS; (e) B. Breit, Angew. Chem., Int. Ed., 2005, 44, 6816–6825 CrossRef CAS PubMed, for important recent contributions, see: (f) V. Agabekov, W. Seiche and B. Breit, Chem. Sci., 2013, 4, 2418–2422 RSC; (g) U. Gellrich, J. Huang, W. Seiche, M. Keller, M. Meuwly and B. Breit, J. Am. Chem. Soc., 2011, 133, 964–975 CrossRef CAS PubMed; (h) U. Gellrich, W. Seiche, M. Keller and B. Breit, Angew. Chem., Int. Ed., 2012, 51, 11033–11038 CrossRef CAS PubMed; (i) A. T. Straub, M. Otto, I. Usui and B. Breit, Adv. Synth. Catal., 2013, 355, 2071–2075 CrossRef CAS.
- http://www.incatt.nl/ .
- (a) D. L. Pompliano, A. Peyman and J. R. Knowles, Biochemistry, 1990, 29, 3186 CrossRef CAS; (b) S. Lüdtke, P. Neumann, K. M. Erixon, F. Leeper, R. Kluger, R. Ficner and K. Tittmann, Nat. Chem., 2013, 5, 762–767 CrossRef PubMed; (c) M. Fujihashi, T. Ishida, S. Kuroda, L. P. Kotra, E. F. Pai and K. Miki, J. Am. Chem. Soc., 2013, 135, 17432–17443 CrossRef CAS PubMed; (d) N. Wu, Y. R. Mo, J. L. Gao and E. F. Pai, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2017–2022 CrossRef CAS PubMed; (e) W. J. Albery and J. R. Knowles, Angew. Chem., Int. Ed. Engl., 1977, 16, 285–293 CrossRef CAS PubMed.
- For reviews, see: (a) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450–2494 CAS; (b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed, and selected examples: (d) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed; (e) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2009, 327, 315–319 CrossRef PubMed.
- (a) R. Breslow, X. Zhang and Y. Huang, J. Am. Chem. Soc., 1997, 119, 4535–4536 CrossRef CAS; (b) J. Yang and R. Breslow, Angew. Chem., Int. Ed., 2000, 39, 2692–2694 CrossRef CAS; (c) J. Yang, B. Gabriele, S. Belvedere, Y. Huang and R. Breslow, J. Org. Chem., 2002, 67, 5057–5067 CrossRef CAS PubMed.
- Z. Fang and R. Breslow, Org. Lett., 2006, 8, 251–254 CrossRef CAS PubMed.
- (a) S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941–1943 CrossRef CAS PubMed; (b) S. Das, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 15911–15913 CrossRef PubMed, see also: (c) R. H. Crabtree, New J. Chem., 2011, 35, 18–23 RSC; (d) S. Das, G. W. Brudvig and R. H. Crabtree, Chem. Commun., 2008, 413–424 RSC.
- J. F. Hull, E. L. O. Sauer, C. D. Incarvito, J. W. Faller, G. W. Brudvig and R. H. Crabtree, Inorg. Chem., 2009, 48, 488–495 CrossRef CAS PubMed.
- (a) P. Fackler, C. Berthold, F. Voss and T. Bach, J. Am. Chem. Soc., 2010, 132, 15911–15913 CrossRef CAS PubMed; (b) P. Fackler, S. M. Huber and T. Bach, J. Am. Chem. Soc., 2012, 134, 12869–12878 CrossRef CAS PubMed.
- F. Voss, E. Herdtweck and T. Bach, Chem. Commun., 2011, 47, 2137–2139 RSC.
- T. Hoke, E. Herdtweck and T. Bach, Chem. Commun., 2013, 49, 8009–8011 RSC.
- A. Bauer, F. Westkamper, S. Grimme and T. Bach, Nature, 2005, 436, 1139–1140 CrossRef CAS PubMed.
- (a) C. Muller, A. Bauer and T. Bach, Angew. Chem., Int. Ed., 2009, 48, 6640–6642 CrossRef PubMed; (b) C. Müller, M. M. Maturi, A. Bauer, M. C. Cuquerella, M. A. Miranda and T. Bach, J. Am. Chem. Soc., 2011, 133, 16689–16697 CrossRef PubMed.
- S. C. Coote and T. Bach, J. Am. Chem. Soc., 2013, 135, 14948–14951 CrossRef CAS PubMed.
- (a) T. Šmejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 311–315 CrossRef PubMed; (b) T. Šmejkal, D. Gribkov, J. Geier, M. Keller and B. Breit, Chem.–Eur. J., 2010, 16, 2470–2478 CrossRef PubMed.
- T. Šmejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 3946–3949 CrossRef PubMed.
- (a) L. Diab, T. Šmejkal, J. Geier and B. Breit, Angew. Chem., Int. Ed., 2009, 48, 8022–8026 CrossRef CAS PubMed; (b) L. Diab, U. Gellrich and B. Breit, Chem. Commun., 2013, 49, 9737–9739 RSC; (c) D. Fuchs, G. Rousseau, L. Diab, U. Gellrich and B. Breit, Angew. Chem., Int. Ed., 2012, 51, 2178–2182 CrossRef CAS PubMed.
- (a) P. Dydio, W. I. Dzik, M. Lutz, B. de Bruin and J. H. N. Reek, Angew. Chem., Int. Ed., 2011, 50, 396–400 CrossRef CAS PubMed; (b) P. Dydio, R. J. Detz and J. N. H. Reek, J. Am. Chem. Soc., 2013, 135, 10817–10828 CrossRef CAS PubMed.
- P. Dydio, M. Ploeger and J. N. H. Reek, ACS Catal., 2013, 3, 2939–2942 CrossRef CAS.
- P. Dydio and J. H. N. Reek, Angew. Chem., Int. Ed., 2013, 52, 3878–3882 CrossRef CAS PubMed.
- (a) P. W. N. M. van Leeuwen and C. Claver, Rhodium Catalyzed Hydroformylation, Kluwer Academic Publishers, Dordrecht, 2000, pp. 29–31 Search PubMed; (b) S. Yu, Y. Chie, Z. Guan, Y. Zou, W. Li and X. Zhang, Org. Lett., 2009, 11, 241–244 CrossRef CAS PubMed; (c) E. R. Nelsen and C. R. Landis, J. Am. Chem. Soc., 2013, 135, 9636–9639 CrossRef CAS PubMed.
- (a) D. B. Grotjahn, C. D. Incarvito and A. L. Rheingold, Angew. Chem., Int. Ed., 2001, 40, 3884–3887 CrossRef CAS; (b) D. B. Grotjahn and D. A. Lev, J. Am. Chem. Soc., 2004, 126, 12232–12233 CrossRef CAS PubMed; (c) D. B. Grotjahn, Chem.–Eur. J., 2005, 11, 7146–7153 CrossRef CAS PubMed.
- (a) D. B. Grotjahn, V. Miranda-Soto, E. J. Kragulj, D. A. Lev, G. Erdogan, X. Zeng and A. L. Cooksy, J. Am. Chem. Soc., 2008, 130, 20–21 CrossRef CAS PubMed; (b) D. B. Grotjahn, E. J. Kragulj, C. D. Zeinalipour-Yazdi, V. Miranda-Soto, D. A. Lev and A. L. Cooksy, J. Am. Chem. Soc., 2008, 130, 10860–10861 CrossRef CAS PubMed.
- P.-A. R. Breuil, F. W. Patureau and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 2162–2165 CrossRef CAS PubMed.
- (a) L. Pignataro, M. Boghi, M. Civera, S. Carboni, U. Piarulli and C. Gennari, Chem.–Eur. J., 2012, 18, 1383–1400 CrossRef CAS PubMed; (b) L. Pignataro, C. Bovio, M. Civera, U. Piarulli and C. Gennari, Chem.–Eur. J., 2012, 18, 10368–10381 CrossRef CAS PubMed.
- P. Dydio, C. Rubay, T. Gadzikwa, M. Lutz and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 17176–17179 CrossRef CAS PubMed.
- (a) W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2010, 132, 10891–10902 CrossRef CAS PubMed; (b) S. Zhu, J. V. Ruppel, H. Lu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2008, 130, 5042–5043 CrossRef CAS PubMed.
- Z. Huang, Z. Chen, L. H. Lim, G. C. P. Quang, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 5807–5812 CrossRef CAS PubMed.
- I. Usui, S. Schmidt, M. Keller and B. Breit, Org. Lett., 2008, 10, 1207–1210 CrossRef CAS PubMed.
- T. Šmejkal and B. Breit, Organometallics, 2007, 26, 2461–2464 CrossRef.
- L. Du, P. Cao, J. Xing, Y. Lou, L. Jiang, L. Li and J. Liao, Angew. Chem., Int. Ed., 2013, 52, 4207–4211 CrossRef CAS PubMed.
- (a) C. U. Grünanger and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 7346–7349 CrossRef PubMed; (b) C. U. Grünanger and B. Breit, Angew. Chem., Int. Ed., 2010, 49, 967–970 CrossRef PubMed; (c) Y. Ueki, H. Ito, I. Usui and B. Breit, Chem.–Eur. J., 2011, 17, 8555–8558 CrossRef CAS PubMed; (d) I. Usui, K. Nomura and B. Breit, Org. Lett., 2011, 13, 612–615 CrossRef CAS PubMed; (e) T. E. Lightburn, M. T. Dombrowski and K. L. Tan, J. Am. Chem. Soc., 2008, 130, 9210–9211 CrossRef CAS PubMed; (f) A. D. Worthy, M. M. Gagnon, M. T. Dombrowski and K. L. Tan, Org. Lett., 2009, 11, 2764–2767 CrossRef CAS PubMed; (g) X. Sun, K. Frimpong and K. L. Tan, J. Am. Chem. Soc., 2010, 132, 11841–11843 CrossRef CAS PubMed; (h) A. D. Worthy, C. L. Joe, T. E. Lightburn and K. L. Tan, J. Am. Chem. Soc., 2010, 132, 14757–14759 CrossRef CAS PubMed; (i) T. E. Lightburn, O. A. De Paolis, K. H. Cheng and K. L. Tan, Org. Lett., 2011, 13, 2686–2689 CrossRef CAS PubMed; (j) C. L. Joe and K. L. Tan, J. Org. Chem., 2011, 76, 7590–7596 CrossRef CAS PubMed, see also: (k) C. S. Yeung and V. M. Dong, Angew. Chem., Int. Ed., 2011, 50, 809–812 CrossRef CAS PubMed.
- (a) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112–114 CrossRef CAS; (b) R. B. Bedford and M. E. Limmert, J. Org. Chem., 2003, 68, 8669–8682 CrossRef CAS PubMed; (c) L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS; (d) M. C. Carrion and D. J. Cole-Hamilton, Chem. Commun., 2006, 4527–4529 RSC.
- A. Iraqi, N. R. Fairfax, S. A. Preston, D. C. Cupertino, D. J. Irvine and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1991, 1929–1935 RSC.
| This journal is © The Royal Society of Chemistry 2014 |