 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalysed Z-selective cross metathesis of allylic-substituted olefins†
Brendan L.
Quigley
and
Robert H.
Grubbs
*
Arnold and Mabel Beckman Laboratories of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: rhg@caltech.edu
First published on 30th October 2013
Abstract
The Z-selective cross metathesis of allylic-substituted olefins is explored with recently developed ruthenium-based metathesis catalysts. The reaction proceeds with excellent stereoselectivity for the Z-isomer (typically >95%) and yields of up to 88% for a variety of allylic substituents. This includes the first synthesis of Z-α,β-unsaturated acetals by cross metathesis and their elaboration to Z-α,β-unsaturated aldehydes. In addition, the reaction is tolerant of a variety of cross partners, varying in functionality and steric profile.
Introduction
Transition metal-catalysed olefin metathesis is a powerful tool for the synthesis of carbon–carbon double bonds in a wide variety of applications.1 However, a major limitation of metathesis has been the lack of a method to selectively generate the Z-olefin product.2 The recent advent of metathesis catalysts that have a preference for the Z-isomer has, for the first time, allowed Z-alkenes to be produced in a general manner.3,4 Though high Z-selectivity has been achieved, the broad reactivity profile characteristic of previous generations of metathesis catalysts is still under development.Ruthenium-based catalysts that contain a chelating NHC ligand, such as 1 and 2 (Fig. 1), are one class of the recently developed Z-selective metathesis catalysts.4,5 In these catalysts, the chelating N-adamantyl substituent6 and bidentate nitrato ligand7 were found to be key to achieving high Z-selectivity across a broad range of reactions, including ethenolysis,8 macrocyclic ring-closing metathesis,9 ring-opening metathesis polymerization,10 asymmetric ring-opening cross metathesis,11 as well as in the more broadly applicable cross metathesis (CM) of two terminal olefins.4,5,12,13 In particular, catalyst 2 has demonstrated Z-selectivities of >95% while achieving turnover numbers of ∼7000 in cross metathesis of unhindered terminal olefins.5 In contrast, allylic-substituted olefins represent a more challenging class of substrates for Z-selective cross metathesis. Allylic substitution introduces increased steric bulk, which can further destabilize the cis-conformation over the trans-conformation of the carbon–carbon double bond.14 Notably, this results in highly selective formation of the E-product (often >90%) with previous generations of Ru metathesis catalysts such as 3 and 4.15,16 While cross metathesis of some allylic-substituted olefins has been very recently achieved with Z-selective Mo-based catalysts (e.g.5 and 6),17,18 this has not been explored with Z-selective Ru-based catalysts. Previous results have demonstrated that substrates with allylic substitution undergo negligible conversion in homodimerization reactions due to the high strain of the products;4,13 however, their reactivity in hetero-cross metathesis has not been explored.19 Ru-based metathesis catalysts in general show broad functional group tolerance and are comparatively air and water stable, lending them to many applications.16,20 Achieving cross metathesis of allylic-substituted olefins represents an important and essential advance in expanding the reactivity profile of Z-selective Ru-based metathesis catalysts.
 | ||
| Fig. 1 Ruthenium- and molybdenum-based metathesis catalysts. | ||
Vinyl acetals were identified as important allylic-substituted olefins which had not been explored in Z-selective metathesis with either Ru- or Mo/W-based catalysts. The products afforded, namely Z-α,β-unsaturated acetals, also serve as precursors to the corresponding Z-α,β-unsaturated aldehydes. E-α,β-Unsaturated aldehydes are available utilizing earlier generations of metathesis catalysts.15 Both classes of compounds are of significant interest for a variety of applications. A number of naturally occurring Z-α,β-unsaturated aldehydes contribute to the scents of various plants and are therefore of interest to the fragrance industry.21 These functionalities also occur in pheromones of several insect species and are potentially of use for mating-disruption as an environmentally friendly alternative to pesticides.22 In both plants and animals, Z-α,β-unsaturated aldehydes are produced during metabolism and their relative abundance is regulated by a number of external and internal stimuli. As a result, some cancer detection and food quality assays rely on these compounds as key indicators.23Z-α,β-Unsaturated acetals and aldehydes also have significant synthetic utility, resulting in their appearance as key intermediates in the synthesis of a number of natural products.24 Furthermore, α,β-unsaturated acetals have been extensively explored as substrates for a number of base- and metal-promoted rearrangements.25 However, while Z-α,β-unsaturated acetals and aldehydes are much sought after, general, broad methods for their synthesis are rare.
Reagents have been developed for the two-carbon homologation of aldehydes to the corresponding α,β-unsaturated compounds, but achieving high Z-stereoselectivity is often challenging and unpredictable.26 This results in the employment of less efficient, multi-step methods.27 For example, the Still–Gennari modification of the Horner–Wadsworth–Emmons reaction28 affords the Z-α,β-unsaturated ester, which can then be reduced to the allylic alcohol and oxidized to afford the aldehyde. In an alternative strategy, alkynyl acetals can be generated by cross-coupling or alkylation, subsequent semi-hydrogenation (typically with Lindlar's catalyst) and deprotection to yield the desired product. Z-Selective metathesis represents an attractive route to α,β-unsaturated acetals and aldehydes that overcomes several shortcomings of the abovementioned methods. Previous methods necessitate the use of strong bases, sensitive organometallic complexes or redox reagents resulting in extensive functional group protections of complex molecules.24,26–28 In contrast, Z-selective metathesis is a more direct method with broad functional group tolerance, reducing the need for protecting groups. In addition, both starting materials are readily available/accessible: vinyl acetals can be efficiently prepared from acrolein29 and cross partners can be sourced from the vast olefin chemical feedstock.
Results and discussion
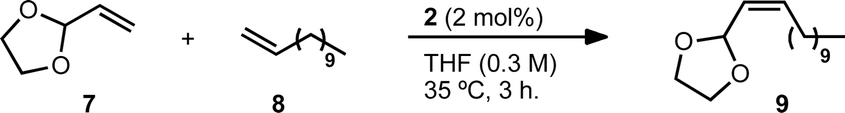
We selected vinyl acetal 7 to explore the optimization of the reaction conditions (Table 1). Acetal 7 is commercially available and the 1,3-dioxolane derivative has enhanced stability to silica gel over acyclic acetals, making it one of the most commonly employed carbonyl protecting groups.30 1-Dodecene (8) was chosen as a cross partner due to its low volatility and known homodimerization by 1 and 2.5,13 Initial conditions utilised 6 equivalents of 8 and 5 mol% of catalyst 2 (entry 1). Under these conditions, product 9 was generated in good yield and high Z-selectivity, with maximum conversion reached at 3 hours. A reduction in the equivalents of 8 and a decrease in catalyst loading were then examined (entries 2–4). Four equivalents of terminal olefin and 2 mol% of catalyst were found to be optimal, giving a similar yield and slightly improved Z-selectivity to initial conditions. Lowering the concentration (entry 5) led to a significant increase in yield, generating product 9 in 92% yield and with 94% Z-selectivity. It is worth noting that across the variety of reaction conditions explored, the Z-selectivity remained consistently high. Further reductions in the excess of terminal olefin and lowering of catalyst loading were both viable but led to longer reaction times and a slight reduction in yield (entries 6–9). In order to demonstrate the versatility of this method, we also explored conditions in which 8 was used as the limiting reagent (Table 2, entries 1–2). Although longer reaction times were required, using 4 equivalents of 7 was found to give comparable yield of product with high Z-selectivity. As in the case of excess 8 (entries 3–4), using 2 equivalents resulted in a lower yield.|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 (mol%) | 7 Equiv. | Conc. (M) | Yielda (%) | Z-Selectivitya (%) |
| a Yield and Z-selectivity determined by GC using tridecane as an internal standard; average of two experiments. b Value at 7 hours. | |||||
| 1 | 5 | 6 | 0.5 | 84 | 93 |
| 2 | 5 | 4 | 0.5 | 87 | 94 |
| 3 | 5 | 2 | 0.5 | 80 | 95 |
| 4 | 2 | 4 | 0.5 | 83 | 95 |
| 5 | 2 | 4 | 0.3 | 92 | 94 |
| 6 | 2 | 2 | 0.5 | 80 | 95 |
| 7 | 2 | 2 | 1.0 | 66 | 95 |
| 8 | 2 | 2 | 0.3 | 82 [87]b | 95 [94]b |
| 9 | 1 | 2 | 0.5 | 74 | 95 |

Under the optimized conditions, we explored reactivity of a number of commercially available Ru-based metathesis catalysts including Z-selective catalyst 1, as well as the commonly employed bis-phosphine catalyst 3 and NHC-substituted catalyst 4. Notably, catalyst 2 generated product 9 in 94% Z-selectivity (Table 3, entry 2). This is in strong contrast to catalyst 1, the previously best Z-selective Ru catalyst, which only achieved 76% selectivity for the Z-isomer (entry 1), clearly highlighting the improved selectivity of catalyst 2 over 1. Both Z-selective catalysts complement previous generations of Ru-complexes (3 and 4), which yield the more thermodynamically stable E-olefin as the major product in comparable yield (entries 3 and 4). It is worth noting that both isomers of product 9 can be generated in high yield and selectivity with Ru-based metathesis catalysts.

The reaction substrate scope was investigated on a preparative scale (0.8–1.0 mmol) with catalyst 2 and we were pleased to find these conditions were applicable to a variety of vinyl acetals (Table 4).31 Vinyl dioxolane cross product (9) was afforded in 82% isolated yield and with >95% Z-selectivity. Other five and six-membered acetal substrates (10–12) performed consistently well, yielding the desired product in 79–85% yield and >95% Z-selectivity. In the case of dimeric acetal 13, the Z,Z-product is afforded as the major isomer (89%) and the Z,E-isomer is the only other observed product (11%). This is consistent with the statistical outcome of two independent steps of ∼94% Z-selectivity.32 The acyclic diethyl acetal afforded cross product 14 in high Z-selectivity, albeit at a slightly reduced yield. Attempts to extend this methodology to related olefins with quaternary allylic substitution (vinyl ketals and orthoesters) have so far been met with limited success.
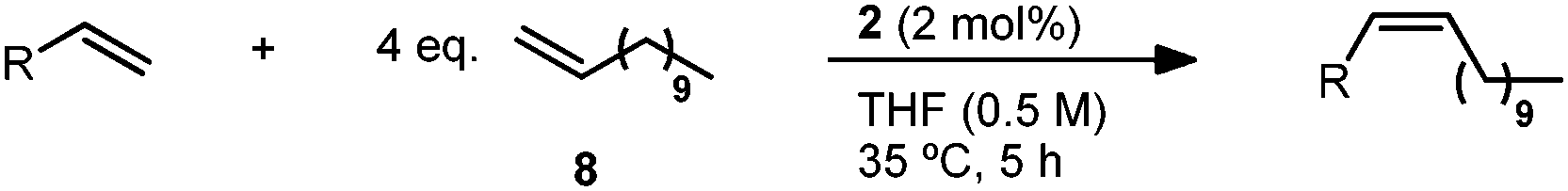

In order to provide further insight into reactivity, we decided to probe a variety of other substrates with related allylic functionalities. Vinyl pinacol boronate has previously been shown to afford highly trans cross products with catalyst 3 (typically, >95% E).15,33 Such products are of use for subsequent Suzuki cross-coupling reactions, where olefin geometry can be efficiently transferred.34 Here, Z-cross product 15 could be generated in good yield (81%) and high Z-selectivity (92%). This result compares favourably with a report by Schrock, Hoveyda and co-workers, except here the typically less expensive terminal olefin can be used in excess.17 Additionally, we investigated 2-vinyloxirane as a substrate for this reaction. Though cross product 16 could be formed in excellent Z-selectivity, the reaction proceeded in only moderate yield.35
The reactivity of vinyl cyclopentane, the all-carbon analogue of substrate 7, was then examined. In this case, methyl-10-undecenoate was used in order to facilitate separation of the cross product from the homodimer of the terminal olefin. While Z-selectivity of product 17 was good (94%), the yield was significantly lower than that for 9 (44% vs. 82%). Increased catalyst loading or gradual addition of vinyl cyclopentane were unsuccessful in restoring the high yields observed in the case of the acetal. Furthermore, vinyl cyclohexane, which is more sterically demanding, demonstrated vastly diminished reactivity, with only minimal conversion after 5 hours.
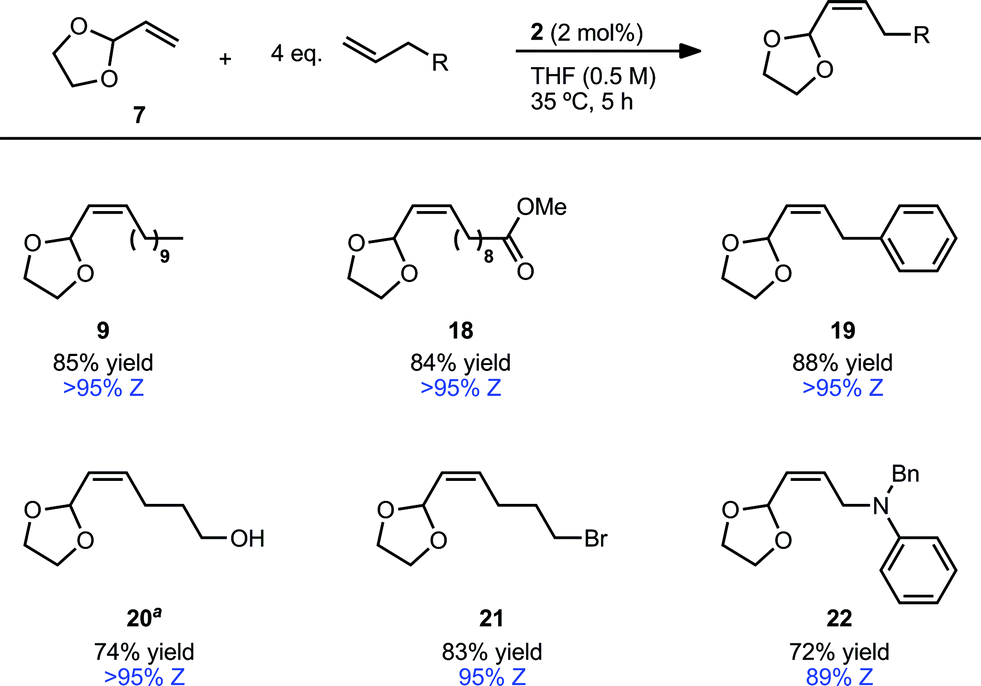
An important factor in achieving a synthetically useful Z-selective CM methodology is tolerance of functional groups that are present in complex molecules. Reaction conditions were found to be compatible with a variety of terminal olefins, which formed cross products with 7 in 72–88% yield and 89–>95% Z-selectivity (Table 5). Notably, several moieties useful for further functionalization could be incorporated, including unprotected alcohols (20)36 and alkyl bromides (21). Though cross metathesis with N-allylaniline resulted in a reduced yield (18% yield, 94% Z), reactivity was restored on protection with a benzyl group. The increased steric bulk, however, likely resulted in the slightly reduced Z-selectivity noted for product 22 (72% yield, 89% Z). Allyl pinacol boronate, a moiety useful for subsequent stereospecific allylation,17,37 was also found to be compatible with the reaction conditions generating product 23 in >95% Z-selectivity (Scheme 1). In order to facilitate purification, 23 was converted to Z-allylic alcohol 24, which was obtained in good yield and high Z-selectivity over the two steps (63% yield, >95% Z).
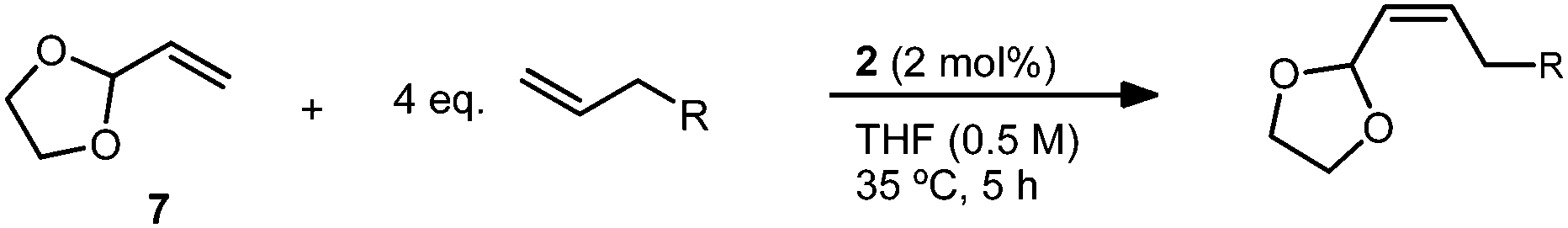
 | ||
| Scheme 1 CM of vinyl dioxolane (7) and allyl pinacol boronate and one-pot conversion to allylic alcohol. | ||
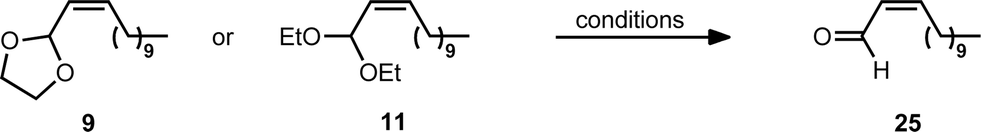
One of the most important uses of Z-α,β-unsaturated acetals is as precursors to the corresponding Z-α,β-unsaturated aldehydes. However, achieving stereo-retentive deprotection is non-trivial and there are only a handful of previous examples in the literature.27b,38 Therefore, a number of conditions were evaluated for deprotection of acetals without loss of Z-stereoselectivity (Table 6). Alkenyl acetals 9 and 11 were selected as they contain two of the most commonly utilised acetal functionalities.30 Deprotection could be effected with Brønsted acid for both 9 and 11 using previously reported reagents (entries 1 and 2).39 Additionally, LiBF4,40 which had not been previously utilised for deprotection of Z-α,β-unsaturated acetals, could also effect the deprotection with excellent retention of alkene stereochemistry (entries 3 and 4).41 This mild method of cleaving the acetal has been demonstrated to be compatible with a wide variety of functional groups.40,42 This would allow incorporation of the more-stable Z-α,β-unsaturated acetal as a masked aldehyde that can be deprotected when needed, lending this methodology to the synthesis of complex molecules.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Subst. | Reagents | Z-Selectivity (%) | Yield (%) | |
| Initial | Final | ||||
a SiO2 2.5 g mmol−1 with 9/11; 5% aq. oxalic acid 10% w/w with SiO2; DCM (0.05 M); r.t., 10 min.
b 1.3 eq. LiBF4; 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 MeCN:H2O (0.1 M); r.t. 10 min. :3 MeCN:H2O (0.1 M); r.t. 10 min.
|
|||||
| 1 | 9 | SiO2, oxalic acida | >95 | >95 | Quant. |
| 2 | 11 | SiO2, oxalic acida | >95 | >95 | Quant. |
| 3 | 9 | LiBF4b | >95 | >95 | 95 |
| 4 | 11 | LiBF4b | >95 | >95 | 92 |
Conclusions
In summary, we have demonstrated that a single ruthenium-based metathesis catalyst can effect the Z-selective cross metathesis of a variety of allylic-substituted olefins with stereoselectivity for the Z-olefin typically in excess of 95%. In doing so, we have developed a mild method for the synthesis of Z-α,β-unsaturated acetals and aldehydes that delivers excellent stereoselectivity and is compatible with a variety of functional groups. Further exploration of reactivity with sterically hindered olefins in tandem with continued development of new catalysts offers the potential to afford Z-olefins with the broad reactivity profile exhibited by previous generations of ruthenium metathesis catalysts.Acknowledgements
This work was financially supported by the NIH (R01GM031332). NMR spectra were obtained on instruments funded by the NIH (RR027690). Materia Inc. is gratefully acknowledged for donation of metathesis catalysts 1–4. S. M. Bronner, J. S. Cannon, M. B. Herbert and V. M. Marx are thanked for helpful discussion.Notes and references
- (a) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2009, 110, 1746–1787 CrossRef PubMed; (b) R. R. Schrock, Chem. Rev., 2009, 109, 3211–3226 CrossRef CAS PubMed; (c) J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS; (d) J. B. Binder and R. T. Raines, Curr. Opin. Chem. Biol., 2008, 12, 767–773 CrossRef CAS PubMed; (e) M. S. Trimmer, in Handbook of Metathesis, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2008, vol. 3, pp. 407–418 Search PubMed; (f) J. Prunet, Curr. Top. Med. Chem., 2005, 5, 1559–1577 CrossRef CAS PubMed.
- D. Lee, Nature, 2011, 471, 452–453 CrossRef CAS PubMed.
- (a) M. M. Flook, A. J. Jiang, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7962–7963 CrossRef CAS PubMed; (b) I. Ibrahem, M. Yu, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3844–3845 CrossRef CAS PubMed; (c) R. K. M. Khan, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2013, 135, 10258–10261 CrossRef CAS PubMed; (d) G. Occhipinti, F. R. Hansen, K. W. Törnroos and V. R. Jensen, J. Am. Chem. Soc., 2013, 135, 3331–3334 CrossRef CAS PubMed.
- (a) K. Endo and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 8525–8527 CrossRef CAS PubMed; (b) B. K. Keitz, K. Endo, P. R. Patel, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 693–699 CrossRef CAS PubMed.
- L. E. Rosebrugh, M. B. Herbert, V. M. Marx, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 1276–1279 CrossRef CAS PubMed.
- For examples of previous non-chelated Ru-based metathesis catalysts with N-adamantyl NHCs see: (a) M. B. Dinger, P. Nieczypor and J. C. Mol, Organometallics, 2003, 22, 5291–5296 CrossRef CAS; (b) M. Mayr, B. Mayr and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2001, 40, 3839–3842 CrossRef CAS; (c) M. R. Buchmeiser, Biorg. Med. Chem. Lett., 2002, 12, 1837–1840 CrossRef CAS.
- Previously nitrato ligands have been shown to decrease activity for other Ru-based metathesis catalysts: (a) M. Jović, S. Torker and P. Chen, Organometallics, 2011, 30, 3971–3980 CrossRef; (b) M. R. Buchmeiser, I. Ahmad, V. Gurram and P. S. Kumar, Macromolecules, 2011, 44, 4098–4106 CrossRef CAS.
- H. Miyazaki, M. B. Herbert, P. Liu, X. Dong, X. Xu, B. K. Keitz, T. Ung, G. Mkrtumyan, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 5848–5858 CrossRef CAS PubMed.
- V. M. Marx, M. B. Herbert, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 94–97 CrossRef CAS PubMed.
- (a) B. K. Keitz, A. Fedorov and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 2040–2043 CrossRef CAS PubMed; (b) L. E. Rosebrugh, V. M. Marx, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10032–10035 CrossRef CAS PubMed.
- J. Hartung and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10183–10185 CrossRef CAS PubMed.
- (a) M. B. Herbert, V. M. Marx, R. L. Pederson and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 310–314 CrossRef CAS PubMed; (b) J. S. Cannon and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 9001–9004 CrossRef CAS PubMed.
- B. K. Keitz, K. Endo, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 9686–9688 CrossRef CAS PubMed.
- R. B. Turner, A. D. Jarrett, P. Goebel and B. J. Mallon, J. Am. Chem. Soc., 1973, 95, 790–792 CrossRef CAS.
- H. E. Blackwell, D. J. O'Leary, A. K. Chatterjee, R. A. Washenfelder, D. A. Bussmann and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 58–71 CrossRef CAS.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- E. T. Kiesewetter, R. V. O'Brien, E. C. Yu, S. J. Meek, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2013, 135, 6026–6029 CrossRef CAS PubMed.
- T. J. Mann, A. W. Speed, R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 8395–8400 CrossRef CAS PubMed.
- While this manuscript was in preparation, a single catalytic example was reported: W.-J. Chung, J. S. Carlson, D. K. Bedke and C. D. Vanderwal, Angew. Chem., Int. Ed., 2013, 52, 10052–10055 CrossRef CAS PubMed.
- (a) S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed; (b) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2000, 34, 18–29 CrossRef.
- (a) M. H. Boelens and L. J. van Gemert, Perfum. Flavor., 1987, 12, 31–43 CAS; (b) G. Ohloff, W. Pickenhagen and P. Kraft, Scent and Chemistry, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2012 Search PubMed.
- (a) E. B. Jang, M. S. Siderhurst, R. G. Hollingsworth, D. N. Showalter and E. J. Troyer, Pest Manage. Sci., 2010, 66, 454–460 CAS; (b) P. E. Howse, I. D. R. Stevens and O. T. Jones, Insect Pheromones and their Use in Pest Management, Chapman and Hall, New York, 1998 Search PubMed.
- (a) K. Jobu, C. Sun, S. Yoshioka, J. Yokota, M. Onogawa, C. Kawada, K. Inoue, T. Shuin, T. Sendo and M. Miyamura, Biol. Pharm. Bull., 2012, 35, 639–642 CrossRef CAS PubMed; (b) F. M. Haddada, H. Manai, D. Daoud, X. Fernandez, L. Lizzani-Cuvelier and M. Zarrouk, Food Chem., 2007, 103, 467–476 CrossRef CAS; (c) S. D. Johanningsmeier and R. F. McFeeters, J. Food Sci., 2011, 76, C168–C177 CrossRef CAS PubMed; (d) B. T. Kebede, T. Grauwet, G. Tabilo-Munizaga, S. Palmers, L. Vervoort, M. Hendrickx and A. Van Loey, Food Chem., 2013, 141, 1603–1613 CrossRef CAS PubMed; (e) T. Balboa-Lagunero, T. Arroyo, J. M. Cabellos and M. Aznar, Am. J. Enol. Vitic., 2011, 52, 527–535 CrossRef.
- (a) T. Brodmann, D. Janssen and M. Kalesse, J. Am. Chem. Soc., 2010, 132, 13610–13611 CrossRef CAS PubMed; (b) W. Roush, D. Coffey and D. Madar, J. Am. Chem. Soc., 1997, 119, 11331–11332 CrossRef CAS; (c) I. Paterson, G. J. Florence, K. Gerlach and J. P. Scott, Angew. Chem., Int. Ed., 2000, 39, 377–380 CrossRef; (d) H. J. Bestmann, O. Vostrowsky, H. Paulus, W. Billmann and W. Stransky, Tetrahedron Lett., 1977, 18, 121–124 CrossRef; (e) A. I. Gerasyuto and R. P. Hsung, J. Org. Chem., 2007, 72, 2476–2484 CrossRef CAS PubMed.
- A. Deagostino, C. Prandi and P. Venturello, Curr. Org. Chem., 2003, 7, 821–839 CrossRef CAS.
- (a) T. Cresp, M. Sargent and P. Vogel, J. Chem. Soc., Perkin Trans. 1, 1974, 37–41 RSC; (b) N. Daubresse, C. Francesch and C. Rolando, Tetrahedron, 1998, 54, 10761–10770 CrossRef CAS; (c) J. Nuzillard, A. Boumendjel and G. Massiot, Tetrahedron Lett., 1989, 30, 3779–3780 CrossRef CAS; (d) R. J. Petroski, K. Vermillion and A. A. Cossé, Molecules, 2011, 16, 5062–5078 CrossRef CAS PubMed; (e) G. Wittig and H. Reiff, Angew. Chem., Int. Ed., 1968, 7, 7–14 CrossRef CAS; (f) R. H. Wollenberg, K. F. Albizati and R. Peries, J. Am. Chem. Soc., 1977, 99, 348–352 CrossRef.
- (a) S. J. Shaw, K. F. Sundermann, M. A. Burlingame, D. Zhang, J. Petryka and D. C. Myles, Biorg. Med. Chem. Lett., 2006, 16, 1961–1964 CrossRef CAS PubMed; (b) S. Zhou, T. Xiao, H. Song and X. Zhou, Tetrahedron Lett., 2012, 53, 5684–5687 CrossRef CAS.
- W. Still and C. Gennari, Tetrahedron Lett., 1983, 24, 4405–4408 CrossRef CAS.
- C. Ikeda, R. Braun and B. Sorenson, J. Org. Chem., 1964, 29, 286–290 CrossRef CAS.
- P. G. M. Wuts and T. W. Greene, in Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 2006, pp. 431–532 Search PubMed.
- It was found that 0.5 M and a 5 hour reaction time were optimal for the range of substrates explored.
- It is worth noting that the two isomers were separable by column chromatography, allowing the Z,Z-product to be isolated cleanly.
- R. Hemelaere, F. Carreaux and B. Carboni, J. Org. Chem., 2013, 78, 6786–6792 CrossRef CAS PubMed.
- (a) G. A. Molander and L. A. Felix, J. Org. Chem., 2005, 70, 3950–3956 CrossRef CAS PubMed; (b) E. M. Woerly, J. R. Struble, N. Palyam, S. P. O'Hara and M. D. Burke, Tetrahedron, 2011, 67, 4333–4343 CrossRef CAS PubMed; (c) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- The reaction was conducted at 20 °C in order to minimize loss of the volatile substrate. A less volatile vinyl epoxide has been shown to generate higher yield (ref. 19).
- At 5 hours some Z to E isomerization of the product had occurred. Investigations into the cause of this isomerization are currently in progress.
- S. D. Goldberg and R. H. Grubbs, Angew. Chem., 2002, 114, 835–838 CrossRef.
- (a) T. R. Kelly, Y. Lee and R. J. Mears, J. Org. Chem., 1997, 62, 2774–2781 CrossRef CAS PubMed; (b) D. A. Dias and M. A. Kerr, Org. Lett., 2009, 11, 3694–3697 CrossRef CAS PubMed; (c) S. Cacchi, G. Fabrizi, L. Moro and P. Pace, Synlett, 1997, 1367–1370 CrossRef CAS.
- T. Sakai, K. Kazuhiko, S. Tsuboi, M. Utaka and A. Takeda, Bull. Chem. Soc. Jpn., 1987, 60, 2911–2915 CrossRef CAS.
- B. H. Lipshutz, Synth. Commun., 1982, 12, 267–277 CrossRef CAS.
- Prolonged reaction times led to noticeable isomerization.
- (a) R. E. Ireland and M. D. Varney, J. Org. Chem., 1986, 51, 635–648 CrossRef CAS; (b) M. Bonin, J. Royer, D. S. Grierson and H. P. Husson, Tetrahedron Lett., 1986, 27, 1569–1572 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for all new compounds. See DOI: 10.1039/c3sc52806e |
| This journal is © The Royal Society of Chemistry 2014 |