Sodium hydroxide-assisted growth of uniform Pd nanoparticles on nanoporous carbon MSC-30 for efficient and complete dehydrogenation of formic acid under ambient conditions†
Qi-Long
Zhu
a,
Nobuko
Tsumori
ab and
Qiang
Xu
*a
aNational Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka 563-8577, Japan. E-mail: q.xu@aist.go.jp; Fax: +81-72-751-9629; Tel: +81-72-751-9562
bToyama National College of Technology, 13, Hongo-machi, Toyama 939-8630, Japan
First published on 16th October 2013
Abstract
Highly dispersed Pd nanoparticles (NPs) deposited on nanoporous carbon MSC-30 have been successfully prepared with a sodium hydroxide-assisted reduction approach. The modification by NaOH during the formation and growth of particles results in the well-dispersed ultrafine Pd NPs on carbon. The combination of distinct interaction between metal and support and high dispersion of NPs drastically enhances the catalytic performance of the resulted catalyst, over which the turnover frequency (TOF) for heterogeneously catalyzed decomposition of formic acid (FA) reaches 2623 h−1 at 50 °C with 100% H2 selectivity, the highest value ever reported under ambient conditions, comparable to those acquired from the most active homogeneous catalysts. Even at 25 °C, the complete dehydrogenation of FA with a TOF as high as 750 h−1 can be achieved.
Introduction
Among the various alternative energy strategies, hydrogen, an environmentally attractive energy carrier that has been considered one of the ultimate energy vectors to connect a host of energy sources to diverse end users for “mobile applications”, will enable a secure and clean energy future.1 Controlled storage and release of hydrogen in a safe and efficient way remains one of the most difficult challenges toward the fuel cell based hydrogen economy.2 Recently, formic acid, a product of biomass processing and photocatalytic CO2 reduction with high energy density, non-toxicity and excellent stability, has been identified as a safe and convenient hydrogen carrier.3–7 Notably, formic acid as liquid hydrogen fuel possesses the distinguished advantages of easy recharging and the availability of the current liquid fuel infrastructure for recharging. Hydrogen stored in FA can be released via a catalytic dehydrogenation reaction (HCOOH → H2 + CO2, ΔG298K = −48.8 kJ mol−1).3b However, the undesirable dehydration pathway (HCOOH → H2O + CO, ΔG298K = −28.5 kJ mol−1), which is typically promoted by heating or acidity, should be strictly controlled, as CO impurities are not well tolerated by fuel cells.3 Only gaseous products (H2/CO2) are formed from selective decomposition of FA, endowing itself with an important advantage over other hydrogen carriers, hence preventing the accumulation of by-products which is a limitation for portable use.Although the decomposition of formic acid has been intensely investigated for hydrogen generation, catalysts possessing prominent activity with facile and selective evolution of CO-free H2 gas under ambient conditions are still few.3 Recently, noteworthy advances have been made in the field of FA dehydrogenation with different homogeneous catalysts.4,5 Beller and co-workers developed a series of Ru–phosphine complexes,5 with which an excellent performance for FA decomposition with an initial turnover frequency (TOF) of 3630 h−1 from FA–amine mixtures at 40 °C has been obtained.5a Laurenczy and co-workers reported a specifically designed water-soluble Ru–TPPTS system, which can release a pressurized H2–CO2 mixture from aqueous solution of FA–sodium formate (SF) at temperatures of 70–120 °C.4a,b More recently, Laurenczy, Beller and co-workers reported a highly active iron catalyst system for the liberation of H2 from FA at 80 °C in a solution of FA in environmentally benign propylene carbonate.4h In contrast to the great progress achieved in the FA dehydrogenation with homogeneous systems, the heterogeneously catalyzed dehydrogenation of FA at ambient temperature is not yet well developed.6 Cao and co-workers applied an ultra-dispersed gold catalyst comprising of gold subnanoclusters deposited on zirconia to a FA–amine mixture, affording a TOF value as high as 1590 h−1.6e Recently, supported Pd-based nanocatalysts have been found to be active for the decomposition of FA, while their catalytic activities and selectivities are poor even at elevated temperature up to 90 °C.6b,7 The development of highly active and selective heterogeneous catalysts for the decomposition of FA in the liquid phase under mild conditions is desired urgently for practical use.
In light of the previous work that the interaction between metal and support and high dispersion of metal NPs may be conducive to the catalytic performance of nanocatalysts, the type of support and the method for preparing ultrafine metal NPs are important for promoting the kinetic properties of FA dehydrogenation.6d,e Herein, we report complete and efficient H2 generation without CO contamination from FA in an aqueous FA–SF solution at ambient temperature, catalyzed by a nanoporous carbon supported Pd nanocatalyst which is prepared by a facile sodium hydroxide-assisted reduction method. Surprisingly, the resultant catalyst affords a TOF value as high as 2623 h−1 at 50 °C with 100% H2 selectivity for heterogeneously catalyzed decomposition of formic acid, the highest value ever reported under ambient conditions, comparable to those acquired from the most active homogeneous catalysts. Maxsorb MSC-30, an elaborate nanoporous carbon, was selected as the support not only because of its incredibly large pore size (2.1 nm) and high specific surface area (>3000 m2 g−1),8 but also because of the possible enhanced interaction between the metal and carbon support. The key to obtaining the excellent catalytic properties of Pd/MSC-30 is the ligand exchange of the precursor complex prior to the reduction with NaBH4, which results in the well-dispersed ultrafine Pd NPs. Moreover, SF in the reaction system plays an important role as a catalyst promoter.
Results and discussion
A facile sodium hydroxide-assisted reduction approach was adopted for the preparation of the Pd/MSC-30 nanocatalyst (Scheme 1). Briefly, 100 mg of MSC-30 carbon was suspended in 2.50 mL water, to which 0.30 mL of aqueous K2PdCl4 solution (0.30 M) was added. The resulting aqueous suspension was subsequently sonicated to homogenize the content, and then treated with 0.50 mL of NaOH solution (2 M). Subsequent reduction using NaBH4 under vigorous stirring generated the product of Pd/MSC-30, which served as an efficient catalyst for H2 generation from FA. Catalytic dehydrogenation of FA was initiated by the introduction of aqueous FA–SF solution into the reaction flask containing Pd/MSC-30 catalyst with vigorous shaking. The catalytic performance of the catalysts was evaluated on the basis of the volumetrically measured amount of gas released during the reaction.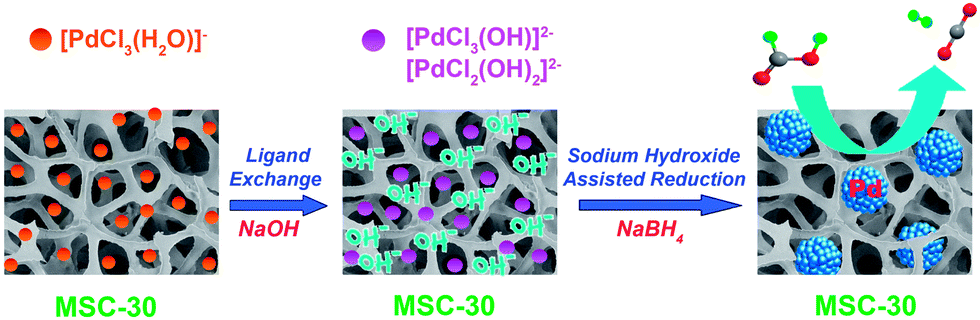 | ||
| Scheme 1 Schematic illustration for Pd/MSC-30 nanocatalyst preparation via a sodium hydroxide-assisted reduction approach and gas (CO2 + H2) evolution from the dehydrogenation of FA–SF. | ||
Remarkably, the Pd/MSC-30 prepared in the presence of base exhibits complete and selective decomposition of FA into H2 and CO2 with an exceedingly high activity. 450 mL of gas without CO detected can be generated within 2.33 min in a FA–SF system at 50 °C (Fig. 1a), which is composed of 440 mL of CO2 + H2 generated from complete FA decomposition and 10 mL of H2 contributed by the hydrolysis of SF (Fig. 1c), indicating complete and efficient conversion of FA into H2 and CO2. The exclusive formation of H2 and CO2 without CO formation has been confirmed by gas chromatography (GC) analyses (Fig. S1 and S2†), which is crucial for fuel cell applications.9 Particularly noteworthy is that the Pd catalyst gave an exceptional turnover frequency (TOF) of 2623 h−1, the highest activity ever reported for heterogeneously catalyzed FA decomposition, even comparable to the most active homogeneous catalysts (Table S1†).3–7,10 The average rate of H2 production was determined to be 684 L H2 h−1 gPd−1, corresponding to a theoretical power density of 923 W h−1 gPd−1 for energy generation.6c,e,f Accordingly, taking an operation efficient of 60% and a typical energy requirement value of 0.5–2.0 W h for portable terminals, one gram of the present Pd/MSC-30 catalyst would be sufficient to supply H2 for 27–110 small proton exchange membrane (PEM) fuel cell devices.6e,11 To be emphasized, few solid catalysts have been reported to be highly active for the decomposition of FA with high selectivity at low temperature.6e Most reported nanocatalysts for FA decomposition usually lose their initially good activities within a short time at the elevated reaction temperature, which may be due to the fact that at elevated temperatures, CO is easier to be evolved to poison the catalysts, and the stability of the catalysts in their size and structure is decreased. To our delight, even at a temperature as low as 25 °C, complete FA decomposition can be readily achieved with TOF of 750 h−1 (Fig. 2). The molar ratio of FA to SF has an obvious effect on the performance of the resulted Pd/MSC-30 catalyst. The activity of the catalyst for decomposition of FA increases with the molar percentage of SF in aqueous FA–SF solution until the value reaches 50%, after which further increase in the percentage of SF has almost no effects on the decomposition of FA (Fig. S9†). In addition, the hydrogen evolution rate greatly depends on the reaction temperature (Fig. 2). The apparent activation energy (Ea) of this process was estimated to be 38.6 kJ mol−1, which is lower than most of those reported for both heterogeneous and homogeneous catalyst systems for FA dehydrogenation.4–6 Thus, FA can be efficiently converted into a CO-free H2-containing stream at convenient temperature by Pd/MSC-30 prepared in an alkaline solution.
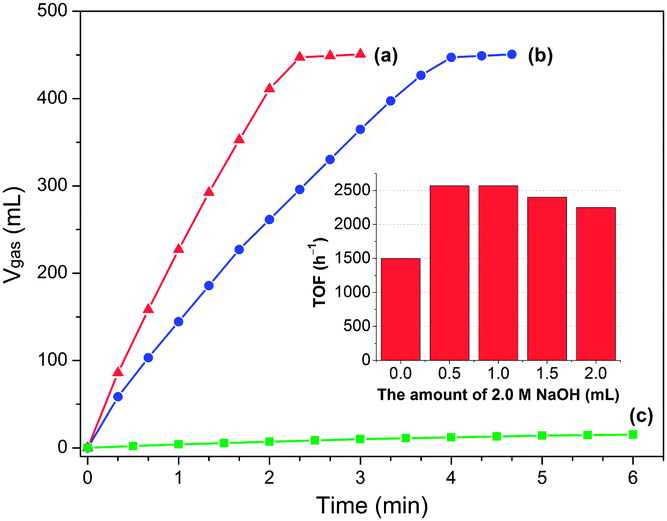 | ||
Fig. 1 Volume of the generated gas (CO2 + H2) versus time for the dehydrogenation of FA–SF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) over Pd/MSC-30 prepared (a) with and (b) without 0.5 mL of 2.0 M NaOH added (nPd/nFA = 0.01, 50 °C), and (c) the hydrolytic dehydrogenation of SF over Pd/MSC-30 prepared with NaOH (nPd/nSF = 0.01, 50 °C). Inset: TOF values of H2 generation from FA–SF over Pd/MSC-30 prepared with different amounts of 2.0 M NaOH added to 2.8 mL precursor solution (K2PdCl4, 0.09 mmol). :1) over Pd/MSC-30 prepared (a) with and (b) without 0.5 mL of 2.0 M NaOH added (nPd/nFA = 0.01, 50 °C), and (c) the hydrolytic dehydrogenation of SF over Pd/MSC-30 prepared with NaOH (nPd/nSF = 0.01, 50 °C). Inset: TOF values of H2 generation from FA–SF over Pd/MSC-30 prepared with different amounts of 2.0 M NaOH added to 2.8 mL precursor solution (K2PdCl4, 0.09 mmol). | ||
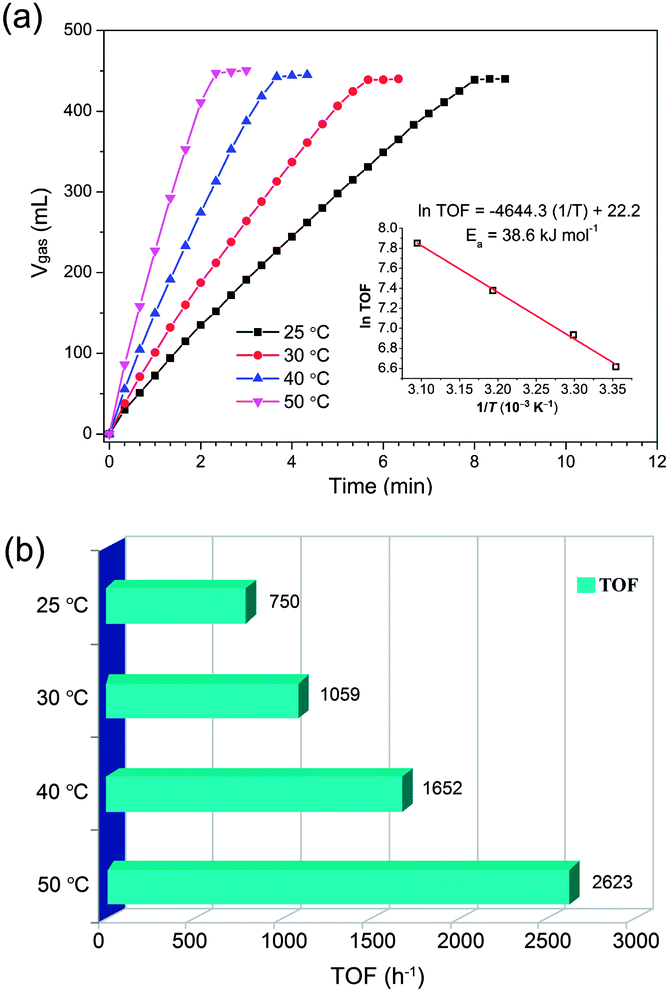 | ||
| Fig. 2 (a) Volume of the generated gas (CO2 + H2) versus time and (b) corresponding TOF values of H2 generation for the dehydrogenation of FA–SF (1:1) at different temperatures over Pd/MSC-30 prepared with NaOH (nPd/nFA = 0.01). Inset of (a): Arrhenius plot (ln(TOF) vs. 1/T). | ||
In addition, the stability of the Pd/MSC-30 catalyst was examined by successively adding aliquots of pure FA (0.34 mL, 9.0 mmol) into the reactor after the completion of previous run, and no significant loss in activity and selectivity was observed over 5 cycles (Fig. S10†). After the catalytic reaction, Pd/MSC-30 shows a similar crystal structure and particle size distribution as that before the reaction (Fig. S14 and S15†). The results reliably indicate that the present catalyst possesses high durability and stability during FA decomposition.
With the absence of NaOH during catalyst preparation, Pd/MSC-30 exhibits a significant decrease in catalytic activity for the decomposition of FA (Fig. 1b), although its apparent activation energy was calculated to be 39.1 kJ mol−1, which is similar to that for Pd/MSC-30 prepared with NaOH (Fig. S12†). It is concluded that the method of catalyst preparation is extremely important for the improvement of kinetics. Further, we have prepared Pd/MSC-30 with various amounts of 2.0 M NaOH solution (0.5 to 2.0 mL) added in 2.8 mL precursor solution (K2PdCl4, 0.09 mmol) and found that the Pd/MSC-30 prepared with 0.5 mL of 2.0 M NaOH exhibited the best catalytic performance in the dehydrogenation of FA (inset in Fig. 1 and S8†), suggesting that a small amount of base during NPs growth is sufficient to improve the catalytic activity of Pd/MSC-30.
As a unique carbon with nanoporous nature, MSC-30 is superior to other kinds of carbon as a support for catalysts. For comparison, Pd NPs supported on another porous carbon, Vulcan XC-72R, with a surface area of 240 m2 g−1 was synthesized in the presence of NaOH. Surprisingly, despite their similar particle size, distribution and crystal structure (vide infra), the catalytic activity of Pd NPs grown on XC-72R is much lower than those on MSC-30 for FA decomposition (Fig. S11†), highlighting the strong interaction between the active NPs and MSC-30. To further determine the effect of the choice of the underlying support on the catalytic performance, we also evaluated Pd NPs supported on Al2O3 nanoparticles (Pd/Al2O3), which showed significantly lower activity under the same condition (Fig. S11†). Based on the above results, it is reliable to believe that the nature of MSC-30 as a support is capable of providing a specific interaction with Pd NPs, which may be the key factor for facilitating the dehydrogenation of FA.
A full characterization of the Pd/MSC-30 catalysts was performed. The transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) images of the Pd/MSC-30 prepared with NaOH show that the Pd NPs are highly dispersed into the framework of MSC-30 with an average particle size of 2.3 ± 0.4 nm based on TEM observations, despite very few aggregated NPs being observed (Fig. 3a, b and S6a†). The corresponding selected area energy dispersion (SAED) pattern indicates the crystalline nature of the Pd NPs (inset in Fig. 3a). In addition, appreciable decreases in the amount of N2 adsorption are observed after the deposition of Pd NPs (Fig. S3†), indicating that the pores of the host framework of MSC-30 are occupied by dispersed Pd NPs and/or blocked by the NPs located on surface as in the case of metal NPs loaded onto porous metal–organic frameworks (MOFs).12 However, when Pd NPs were loaded without NaOH, a larger average particle size of 3.6 ± 0.6 nm was observed (Fig. 3c and S6b†), although the X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) analyses show that the two catalysts possess similar crystalline and electronic structures (Fig. S4 and S5†). Therefore, NaOH serves as an efficient dispersing agent to control the size distribution during the formation of Pd NPs in the present system. It has been known that the catalytic activity generally increases with decreasing metal NP size, as smaller particles possess higher surface areas available for reactants.6g,13 Since there is no significant difference in the crystalline and electronic properties of Pd particles between the two nanocatalysts, it is reasonable to believe that the decrease in particle size of Pd NPs on MSC-30 due to the presence of NaOH should be responsible for the drastic enhancement of the catalytic performance. The highly dispersed Pd NPs on XC-72R and Al2O3 supports with average particle sizes of 2.8 ± 0.5 and 3.3 ± 0.3 nm, respectively (Fig. S7†), further demonstrate that NaOH plays an important role in controlling the growth of small-size Pd NPs.
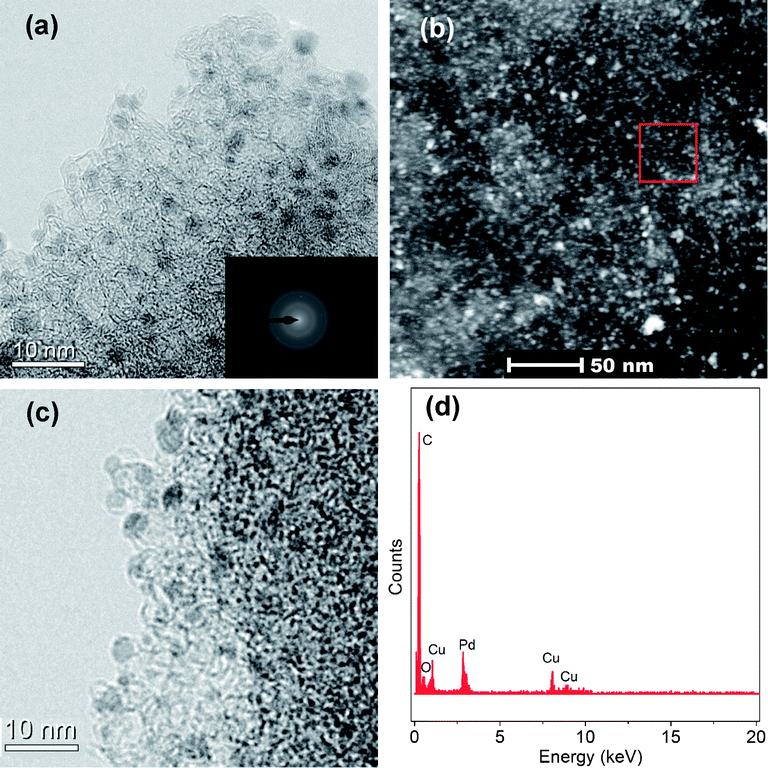 | ||
| Fig. 3 (a) TEM (inset SAED) and (b) HAADF-STEM images of Pd/MSC-30 prepared with NaOH, (c) TEM image of Pd/MSC-30 prepared without NaOH, and (d) EDX pattern of the selected area in (b). The copper signals originate from the TEM grid. | ||
In order to investigate the reason for the change in particle size of Pd NPs induced by NaOH addition, we tried to identify the coordination structure of the precursor in the absence and presence of NaOH by using UV-Vis absorption spectroscopy. The metal precursor in aqueous solution existed as a hydrated ion [PdCl3(H2O)]−, which was confirmed by its ligand-to-metal charge transfer (LMCT) band around 235 nm (Fig. 4).14 After adding an amount of NaOH corresponding to ten equivalents of K2PdCl4 to the precursor aqueous solution, the LMCT band of [PdCl3(H2O)]− complex disappeared immediately, along with the appearance of a broad absorption band centered at 268 nm, which can be assigned to the LMCT band of mixed chlorohydroxypalladium(II) species, e.g., [PdCl3(OH)]2− and [PdCl2(OH)2]2−.15 However, the intensity of the new absorption band was maintained by further addition of aliquots of NaOH, indicating complete ligand exchange (Fig. S13†). Moreover, a noticeable decrease of the 268 nm band can only be observed in the spectra, along with the observation of some precipitation, after a long treatment with alkaline solution. Thus, the formation of colloidal Pd(OH)2 can be excluded during the formation of the Pd NPs on carbon in the presence of NaOH. Consequently, such ligand exchange of Cl− for OH− prior to the reduction with NaBH4 accounts for the decrease in particle size of Pd NPs, which is probably due to the enhanced electronic interaction between the chlorohydroxypalladium(II) species and carbon support. Namely, the precursor complexes containing OH− ligands are much more favorable for the formation of small Pd NPs on the supports than those without OH− ligands.
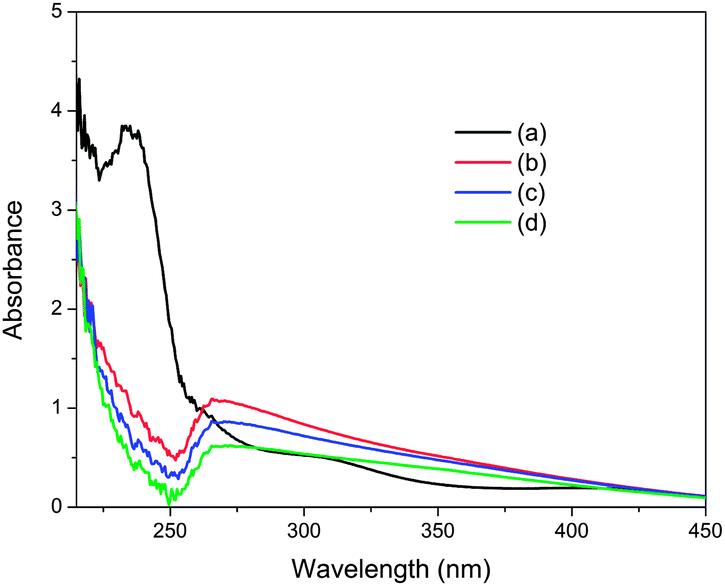 | ||
| Fig. 4 UV-Vis spectra of aqueous K2PdCl4 solution (a) before and (b) 0, (c) 30 and (d) 60 min after the addition of NaOH ([NaOH]/[K2PdCl4] = 10). | ||
Conclusions
In summary, a facile strategy has been successfully applied by using a sodium hydroxide-assisted reduction approach for the preparation of well-dispersed ultrafine Pd NPs, which is induced by the ligand exchange of the precursor complex prior to the reduction with NaBH4. The nanoporous carbon MSC-30 proved to be a distinct and powerful support for the Pd-based catalyst with strong interaction between metal and support. Catalyzed by the resultant Pd/MSC-30, a highly efficient and complete hydrogen generation from FA in a FA–SF system without undesired CO contamination, and with the highest TOF value in the heterogeneously catalyzed FA dehydrogenation, even comparable to those acquired from the most active homogeneous catalysts, was achieved under mild conditions. This readily accessible and highly effective monometallic catalyst is believed to strongly promote the practical development of formic acid as a viable hydrogen storage medium for fuel cells and chemical synthesis applications.Acknowledgements
The authors are thankful to the reviewers for valuable suggestions, Dr Takeyuki Uchida for TEM measurements and AIST and METI for financial support.Notes and references
- (a) L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed; (b) S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246–256 CrossRef CAS PubMed; (c) P. P. Edwards, V. L. Kuznetsov, W. I. David and N. P. Brandon, Energy Policy, 2008, 36, 4356–4362 CrossRef PubMed; (d) H. M. Chen, C. K. Chen, R.-S. Liu, L. Zhang, J. Zhang and D. P. Wilkinson, Chem. Soc. Rev., 2012, 41, 5654–5671 RSC.
- (a) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed; (b) A. M. Seayad and D. M. Antonelli, Adv. Mater., 2004, 16, 765–777 CrossRef CAS; (c) J. Germain, J. M. Fréchet and F. Svec, Small, 2009, 5, 1098–1111 CrossRef CAS PubMed; (d) S.-K. Kim, W.-S. Han, T.-J. Kim, T.-Y. Kim, S. W. Nam, M. Mitoraj, Ł. Piekoś, A. Michalak, S.-J. Hwang and S. O. Kang, J. Am. Chem. Soc., 2010, 132, 9954–9955 CrossRef CAS PubMed; (e) D. Neiner, A. Karkamkar, M. Bowden, Y. Joon Choi, A. Luedtke, J. Holladay, A. Fisher, N. Szymczak and T. Autrey, Energy Environ. Sci., 2011, 4, 4187–4193 RSC; (f) Z.-G. Huang and T. Autrey, Energy Environ. Sci., 2012, 5, 9257–9268 RSC.
- (a) F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef PubMed; (b) S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC; (c) S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS PubMed; (d) S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed; (e) M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC; (f) M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 RSC.
- (a) C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS PubMed; (b) C. Fellay, N. Yan, P. J. Dyson and G. Laurenczy, Chem.–Eur. J., 2009, 15, 3752–3760 CrossRef CAS PubMed; (c) Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC; (d) T. C. Johnson, D. J. Morris and M. Wills, Chem. Soc. Rev., 2010, 39, 81–88 RSC; (e) A. Boddien, B. Loges, F. Gärtner, C. Torborg, K. Fumino, H. Junge, R. Ludwig and M. Beller, J. Am. Chem. Soc., 2010, 132, 8924–8934 CrossRef CAS PubMed; (f) A. Boddien, F. Gärtner, R. Jackstell, H. Junge, A. Spannenberg, W. Baumann, R. Ludwig and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8993–8996 CrossRef CAS PubMed; (g) S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496–1497 CrossRef CAS PubMed; (h) A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed; (i) J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- (a) A. Boddien, B. Loges, H. Junge and M. Beller, ChemSusChem, 2008, 1, 751–758 CrossRef CAS PubMed; (b) B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962–3965 CrossRef CAS PubMed; (c) A. Boddien, B. Loges, H. Junge, F. Gärtner, J. R. Noyes and M. Beller, Adv. Synth. Catal., 2009, 351, 2517–2520 CrossRef CAS.
- (a) Y. Huang, X. Zhou, M. Yin, C. Liu and W. Xing, Chem. Mater., 2010, 22, 5122–5128 CrossRef CAS; (b) X. Gu, Z.-H. Lu, H.-L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS PubMed; (c) K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. Bagot, E. A. Marquis, G. D. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed; (d) S. Jones, J. Qu, K. Tedsree, X. Q. Gong and S. C. E. Tsang, Angew. Chem., Int. Ed., 2012, 51, 11275–11278 CrossRef CAS PubMed; (e) Q.-Y. Bi, X.-L. Du, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 8926–8933 CrossRef CAS PubMed; (f) Z.-L. Wang, J.-M. Yan, Y. Ping, H.-L. Wang, W.-T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed; (g) S. Zhang, Ö Metin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3681–3684 CrossRef CAS PubMed; (h) M. Yadav, T. Akita, N. Tsumori and Q. Xu, J. Mater. Chem., 2012, 22, 12582–12586 RSC.
- M. Yadav, A. K. Singh, N. Tsumori and Q. Xu, J. Mater. Chem., 2012, 22, 19146–19150 RSC.
- S.-I. Jeon, H.-R. Park, J.-G. Yeo, S. Yang, C. H. Cho, M. H. Han and D. K. Kim, Energy Environ. Sci., 2013, 6, 1471–1475 CAS.
- (a) K. V. Kordesch and G. R. Simader, Chem. Rev., 1995, 95, 191–207 CrossRef CAS; (b) F. de Bruijn, Green Chem., 2005, 7, 132–150 RSC.
- (a) Z.-L. Wang, J.-M. Yan, Y. Ping, H.-L. Wang, W.-T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed; (b) S. Zhang, Ö. Metin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3681–3684 CrossRef CAS PubMed.
- H. Tsuchiya and O. Kobayashi, Int. J. Hydrogen Energy, 2004, 29, 985–990 CrossRef CAS PubMed.
- (a) J. Hermannsdörfer, M. Friedrich, N. Miyajima, R. Q. Albuquerque, S. Kümmel and R. Kempe, Angew. Chem., Int. Ed., 2012, 51, 11473–11477 CrossRef PubMed; (b) Q.-L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef CAS PubMed.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed.
- T. Hara, M. Ishikawa, J. Sawada, N. Ichikuni and S. Shimazu, Green Chem., 2009, 11, 2034–2040 RSC.
- T. Harada, S. Ikeda, M. Miyazaki, T. Sakata, H. Mori and M. Matsumura, J. Mol. Catal. A: Chem., 2007, 268, 59–64 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; GC, PXRD, BET, XPS, TEM for catalysts; results of catalytic dehydrogenation of FA; durability and stability results of catalysts. See DOI: 10.1039/c3sc52448e |
| This journal is © The Royal Society of Chemistry 2014 |