
Water film inside graphene nanosheets: electron transfer reversal between water and graphene via tight nano-confinement†
Ruixia Songab,
Wei Fengab,
Camilo A. Jimenez-Cruzc,
Bo Wangab,
Wanrun Jiangab,
Zhigang Wang *ab and
Ruhong Zhou*acd
*ab and
Ruhong Zhou*acd
aInstitute of Atomic and Molecular Physics, Jilin University, Changchun, 130012, China. E-mail: wangzg@jlu.edu.cn; ruhongz@us.ibm.com
bJilin Provincial Key Laboratory of Applied Atomic and Molecular Spectroscopy (Jilin University), Changchun, 130012, China
cComputational Biology Center, IBM Thomas J. Watson Research Center, Yorktown Heights, NY 10598, USA
dInstitute of Quantitative Biology and Medicine, SRMP and RAD-X, Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Soochow University Medical College, Suzhou 215123, China
First published on 24th November 2014
Abstract
Based on quantum dynamics simulations using the density functional tight-binding (DFTB) method, we provide a detailed geometric and electronic structure characterization of a nano-confined water film within two parallel graphene sheets. We find that, when the distance between the graphene bilayer is reduced to 4.5 Å, the O–H bonds of the water molecules become almost parallel to the bilayer; however, further reducing the distance to 4.0 Å induces an abnormal phenomenon characterized by several O–H bonds pointing to the graphene surface. Electronic structure analyses revealed that the charge transfers of these nano-confined water molecules are opposite in these two situations. In the former scenario, the electron loss of each water molecule in the confined aqueous monolayer is approximately 0.008 e, with electrons migrating to graphene from the p orbitals of water oxygen atoms; however, in the latter case, the electron transfer is reversed, with the water monolayer gaining electrons from graphene in excess of 0.017 e per water molecule. This reversed behavior arises as a result of the empty 1s orbitals of H atoms, which are disturbed by the delocalized π orbitals formed by the p electrons of the carbon atoms. Our current study highlights the importance of the nano-confinement on the electronic structures of interfacial water, which can be very sensitive to small changes in physical confinement such as a small reduction in the graphene interlayer distance, and may have implications in de novo design of graphene nano-channels with unique water transport properties for nanofluidic applications.
Introduction
The properties of interfacial water are mainly driven by the van der Waals (vdW) interactions1 comprising primarily of hydrogen bonds,2–4 H–π5 and other comparatively weaker interactions. Interfacial properties are prevalent in many relevant systems such as adsorption,6,7 confinement,8,9 nanopores10 and nano-channels,11,12 and permeation.13 Research into the unique properties of interfacial water (such as wetting, catalysis, lubrication, and the adjustment of hydrophobic collapse in multidomain proteins14–17) is therefore of extreme interest from both scientific and technological standpoints. On the other hand, since the discovery of graphene18 and after many successful advances in manufacturing methodologies,19–22 there has been a growing interest in the study of the solid–liquid interface structures formed by water molecules adsorbed on graphene sheets.6,7,23–25 When a water molecule is intercalated into a bilayer graphene, thus forming a system of graphene–water–graphene (GWG), the constraints of this interlayer conformation strongly affect the adsorption sites and the orientation of the water molecule.26 Interestingly, the interlayer structure may adjust the transport properties of water in graphene nano-channels, by changing the shear stress on the graphene planes.11 This mechanism has been suggested to have application prospects in the design of nanofluidic devices27,28 useful in several fields of manufacture, testing and characterization. In order to successfully affect the physical and/or chemical properties of graphene, interlayer distances in GWG systems are generally approached or even reached the limit of the van der Waals (vdW) radius of a carbon atom.8,29 This suggests that the electronic structures of solid–liquid–solid interface may be changed due to the close distances between molecules. It has been speculated that the properties (geometric, electronic and dynamical) of water confined between two parallel graphene layers,27 are presumably related to subtle rearrangements of electronic structures at the interface with the confining medium.30 However, an in depth understanding of the electronic structures of water–graphene sandwich structure is so far preliminary.Unlike monolayer graphene, multilayer graphenes have unique interlaminar interactions,31 which result in differences in the electron bands varying with the number of layers.32 Specifically, the controlling of band structure suggests the potential application of multi-layer graphenes for switching functions.33,34 The presence of water molecules could not only adjust the interlaminar structure,26 but also reduce the friction between the graphite sheets.35,36 As an example of such phenomena, it has been recently reported how the properties of liquid were affected by confinement, based on classical molecular dynamics simulations.8 Previous studies have also investigated the effects of the interlayer distance on the dynamics of the aqueous system. In this case, the layers were compressed until they reached d ∼ 0.34 nm, followed by an structure relaxation, discussing in this way the phenomena of water crystallization and ion-induced polarization.29 However, to the best of our knowledge, there are no reports of a detailed study of the electrical properties for confinement systems within the range of distances of interest. At such short distances, the hydrogen bonding of water molecules, H–π interactions between water molecules and graphene, etc., may be different from intermolecular weak adsorption. It is therefore necessary to study the structure and dynamical behavior of interlayer water under such a tight confinement near the vdW boundary based on quantum mechanics, with the end goal of revealing possible changes in the electronic structures and their influence on the dynamical and transport properties of the GWG system.
Taking into account the large size of the sandwich configurations, the accuracy, and computational effort required, the density functional tight-binding (DFTB) method was chosen. This method effectively maintains the high accuracy of density functional theory (DFT), while providing a severe increase in performance (almost 103 to 104) when compared with conventional DFT,37 with the additional benefit of allowing a complete analysis of the electronic structure.38,39 Very recently, the third generation of the quantum approximate density functional tight-binding methodology (DFTB3) method developed on the basis of DFTB method, has been used to study the solvent effects39 as well as the adsorption properties of surface water.40 The demonstration on wetting properties of graphene surface based on DFTB3 discussed here agree and help to explain the most recent experimental results.41
In this study, we investigated the structure and dynamics of GWG system for aqueous layer trapped between two parallel graphene sheets based on the DFTB3 method, focusing on the variation of electronic structures with the change of the distance of the graphene bilayer. Our results display an anomalous distribution in water conformations, accompanied with an unusual charge transfer between water and graphene when the distance decreases to near or even below the vdW boundary. All these interesting effects are attributed to the influences of peculiar electronic structures of graphene on the water monolayer under tight confinement.
Theoretical method and model
Given the large size of the system of interest, a detailed density functional theory (DFT) study of the interactions between water and graphene, would prove computationally prohibitive. However, by using the third generation of the quantum approximate density functional tight-binding methodology (DFTB3)42 as a simplified variant of DFT, we could describe appropriately the quantum properties while being tractable computationally.40 In DFTB3, the added γ functions and Hubbard derivatives for hydrogen and oxygen atoms43 result suitable in the studies of such interfacial water systems.40,44 In this study, we performed dynamic simulations of a water film confined between periodic graphene bilayer by using the quantum mechanical DFTB3 method. Slater–Kirkwood dispersion was used in all molecular dynamics simulation and geometry optimization to improve the computational accuracy. This allows us to discuss the impact of different interlayer distances on the orientations of O–H vectors in water molecules, the charge transfer between the water molecules and graphene layers, the bonding nature, the orbital properties, and other related aspects.Here, the GWG system refers a water film confined by a graphene bilayer. The distance between periodic bilayer graphene (of dimensions 12.78 Å × 9.84 Å) is set to be 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 8.0, and 9.0 Å. The initial structures of the single crystal cell containing 18 water molecules between the graphene bilayer were built based on the consensus that water molecules generally form six-membered rings when exposed to graphene, resulting in a water density of 0.143 Å−2 (the stable structures with water densities of 0.072 Å−2 and 0.151 Å−2 are shown in ESI Fig. S1†). Previous studies have shown that the adsorption of water molecules on graphene does not result in obvious bending of the graphene surface.40 Since we are mainly concerned about the effects of the nano-confinement on the properties of the water molecules, the graphene sheets were fixed during both, the dynamics simulation and the structure optimization. Similar model systems have been used in the study of the dynamics of water trapped between hydrophobic solutes45 and parallel graphene sheets.25,46
The equations of the motion of the nuclei were integrated using the Velocity-Verlet algorithm47 under an NVT ensemble via the Andersen thermostat at 300 K.48 All molecular dynamics simulations are performed with a time step of 1 fs during 100 ps. The atomic spatial location and velocity distribution at the end of the simulation at 300 K was then used as the initial configuration for the relaxation 500 K to validate the conclusions of geometric relaxation and dynamical equilibrium for the systems (see ESI†). Each system is sampled five times or more to ensure meaningful statistics. We analyzed density distributions of O and H atoms, charge transfer profiles and average Mermin free energy for each system. Complementing the results of the molecular dynamics simulation, and in order to properly understand the impact of the interlayer distance on the electronic properties of the system, we identified five typical conformations with low Mermin free energy during the simulations of 100 ps at 300 K and 500 K, respectively, and used them as templates to carry further geometry optimization.
Results and discussion
We performed a series of quantum dynamics simulations of the GWG system with the different interlayer distances covering the range of 4.0 to 9.0 Å. The results show that the water molecules exhibit ordered distributions at equilibrium as the distance decreases. Typical conformations of the GWG system under dynamic equilibrium (after 100 ps) at 300 K are shown in Fig. 1. When water molecules are constrained by graphene sheets separated by 9.0 or 6.5 Å, the water molecules display a slight stratification and more complex dynamics. When the distance is decreased to 4.5 Å, most of the water O atoms deviate from the center of the six-membered rings, and water molecules are no longer able to form a conventional six-membered ring structure (bottom part of Fig. 1b). The H atoms are now almost parallel to the graphene sheets (Fig. 1a). Interestingly, with only a slight decrease in the distance from 4.5 Å to 4.0 Å, most O atoms relocated above the hole of graphene (top part of Fig. 1b), in contrast to the previous scenario, the H atoms not engaged in hydrogen bonds point to the C–C bonds of graphene. The minimum distance between an H atom and the surface of graphene is 1.12 Å (Fig. 1a). Because the distances between H atoms and graphene are so short, H atoms would not point to the center of the six-membered rings and hardly form any effective O–H⋯π interactions. Since the distance is very small, the water molecules are strongly confined, resulting in a clear trend of breakage of many OH bonds, leading to not being able to hold one layer of water molecules sterically. This resembles a similar phenomenon that has been observed in previous macroscopic theoretical studies of water inside critical double graphite–CH3 plates with the distance of 3–4 Å, when the contact angle is about 100°.49 In the following, we mainly concentrate on the anomalous behaviors of the GWG system under the tight confinement within the vdW boundary. | ||
| Fig. 1 Typical conformations of GWG systems in dynamic equilibrium (100 ps) at 300 K. (a) Side views of the constructed systems with interlayer distances of 4.0, 4.5, 6.5 and 9.0 Å as labeled. (b) Top views of the systems with distances of 4.0 Å and 4.5 Å. The minimum distance between an H atom and the surface of graphene is 1.12 Å. | ||
In order to obtain more details about the influence of different distances on the interfacial water, we further analyzed density distributions of hydrogen and oxygen atoms in water molecules using the trajectory at 300 K. The resulting plots are shown in Fig. 2a and b. For the relatively large distances, in particular for distances greater than 6.0 Å, the distribution areas of O atoms are clearly wide, and the curves tend to flatten. This flattening of the densities indicates the stratification of water molecules along the z axis, with a more dynamic behavior with larger fluctuations observed at larger distances. However, it is noteworthy that the density curves of H and O atoms are not completely symmetrical, especially above 6.0 Å (see arrows in Fig. 2b). It can be seen that water molecules are not completely equally adsorbed in the vicinity of the graphene bilayer, but slightly inclined to one of graphene sheets. This is an effect of the electronic structure that will be discussed below.
 | ||
| Fig. 2 Density distributions of hydrogen (a) and oxygen atoms (b) of confined water molecules, for the different distances studied. Here, the z direction is taken to be perpendicular to the two parallel graphenes, with z = 0 being the middle position between two graphene layers. The hydrogen and oxygen distributions are shown in the range of −3 to +3 Å and −2 to +2 Å, respectively (consistent with previous studies, the water molecules gathered near graphene close to 2.5 Å, which was regarded as the interface region30). (c) Mulliken charge transfer of the GWG system with different distances, black and red represent the average charge transfer in dynamic processes (md) and charge transfer of optimized structure (opt), respectively. Charge transfer value is net charge of each water molecule. | ||
As the distance decreases, the distribution areas of H and O atoms become narrower, leading to significant peaks of the curves. It can be observed that O atoms are located in the central area between bilayer graphene at 4.5 Å. When the distance decreases to 4.0 Å, three main distribution areas in hydrogen atoms are observed, with the distributions on both sides being approximately symmetrical and lower than that of the middle region. According to the typical structures of dynamic processes shown in Fig. 1, this is because the water molecules are squeezed by the graphene sheets, forcing most hydrogen atoms participating in hydrogen bonds to be located at the center of the graphene bilayer, with only a few hydrogen atoms that are non-hydrogen bonded pointing to the graphene surfaces. And similar to the d = 4.5 Å case, O atoms are still located in the central area between graphene bilayer at 4.0 Å. Thus, under the tight confinement below the vdW boundary, O atoms are strictly bound in the middle of the interlayer graphene. This result, in conjunction with previous results on similar systems with confinement distances shorter than the vdW boundary, e.g., the studies of endohedral metallofullerenes50,51 show that it will bring about electronic correlation between internal confinement molecular and the outer shell. Therefore, we put emphasis on the electronic structures of the GWG system within the vdW boundary in this study. At 500 K, density distributions of oxygen and hydrogen atoms are similar with the results of 300 K. However, with the elevated temperature, the dynamics of water molecules become more active, making the distribution curves smoother (see ESI Fig. S2†).
To date, there are no detailed analyses of the changes in electronic structure of the GWG system with the variation of the distance reported in the literature. Hence, here we study the charge transfer, differential charge density, density of states, molecular orbitals, and charge density of the stabilized systems. Mulliken charge is the most important basic component of the Self Consistent Mulliken Charge Density-Functional Tight-Binding Method (SCC-DFTB),52 which is directly and widely used to analyze electronic properties of the surface adsorption,38,40,53 stacking effect,37 hydrolysis reactions,54 imogolite aggregation,55 and so on.
Fig. 2c shows the average charge transfer of the GWG system along the quantum dynamics trajectory and charge transfer based on structure optimization, as an aid to understand the impact of different distances on charge transfer. It can be seen that, while above 6.0 Å there is little or no charge transfer from graphene bilayer to aqueous layer, the charge transfer becomes obvious as the distance is reduced to less than the vdW boundary. Particularly, for d = 4.5 Å, the electronic balance flips sign, with the water layer displaying a net loss of electrons of around 0.008 e (0.012 e for optimized structure) per water molecule. When the distance is decreased to 4.0 Å, the electronic balance flips again, and the overall water layer gains electrons in excess of 0.017 e per water molecule. The above analysis shows that the distance has direct influence on charge transfer of the GWG system, and the charge transfer effects become stronger when the distance is less than the vdW boundary. What is even more, the direction of charge transfer switches at around d = 4.0–4.5 Å. The results of 500 K are contained in the ESI (see Fig. S3†), and the trends are consistent with that of 300 K. Previous studies of low-dimensional carbon materials showed that the degree of confinement was found to have more influence on the interfacial friction, lubrication36 and diffusion27 of systems. And the electronic structure is an important factor in regulating the sandwich confinement structure. The unique charge transfer appeared within the vdW boundary may contribute to the design of new devices with useful applications in nanofluidics.
To further identify the sources of charge transfer and better understand the relation between the conformation and electronic structure, we analyzed charge density and differential charge density (Fig. 3). Combined with the distribution of H and O atoms (Fig. 1), for the longer distances, some hydrogen atoms of the water molecules point to the graphene, and the aqueous layer obtains a minimal amount of electrons. The corresponding distributions of differential charge density are listed in the ESI Fig. S4.† However, for d = 4.5 Å, the direction of charge transfer reverses from the above described situation. This is related to the parallel conformations of water molecules and graphene bilayer, as shown in Fig. 3, where the similar rearrangement of water molecules under the vdW boundary influences their electronic structures. Because the atomic radius of O is significantly larger than that of H,56 O atoms are more limited by the confinement from the graphene bilayer in this case. This overlap of electron clouds between O and graphene, leads to the electronic migration from the p orbitals of oxygen atoms to graphene. That is why the confined water molecules lose electrons to the graphene sheets at distances around 4.5 Å. When the distance is further decreased, it can be readily observed that water molecules are strongly constrained for the distances shorter than 4.0 Å, with the closest distance between a H atom of water and a C atom in graphene only 1.56 Å (marked in Fig. 3a; and a 1.12 Å shortest distance to the graphene surface, marked in Fig. 1a), which leads to a tendency to make covalent bonds. Here the aqueous layer obtains electron in excess of 0.017 e per water molecule (see Fig. 2c), due to the hydrogen atoms (non-hydrogen bonding) pointing toward the graphene bilayer. In this case, the hydrogen atom infiltrates the surface of graphene sheets (illustrated in Fig. 2a and 3), leading to the empty 1s orbital of H atom being disturbed by the delocalized π bonds of p electrons of C atoms in graphene. To further verify the conclusions reached in Fig. 1, we also show dissociation trends of the water molecules in Fig. 3, which is consistent with previous theoretical reports of water ice interphase under high-pressure.9,57 It is then observed that the impact of the distance (under the vdW boundary) between the graphene layers on differential charge density is not only significant, but also may reverse the sign, especially in the 4.0–4.5 Å range. Our results also indicate that the distribution trend of electron densities at 500 K is consistent with that of 300 K (see Fig. S5†). In order to better reflect these overlapping sources in charge density and differential charge density, we analyzed the nature of the frontier orbitals, which shows the orbital overlap between water molecules and graphene sheets. It can even merge into one organic “orbital” in the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) at 4.0 and 4.5 Å.
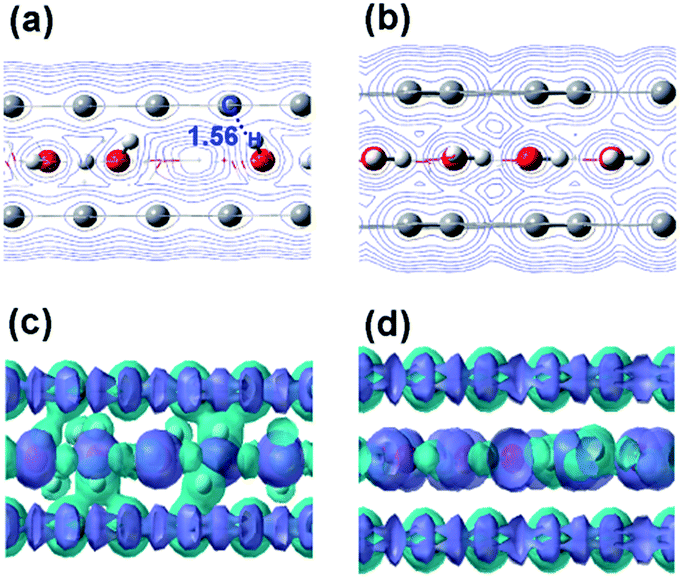 | ||
| Fig. 3 Charge density contours of the GWG system with the distances of 4.0 Å (a), 4.5 Å (b), respectively. Differential charge density of the GWG system with the distances of 4.0 Å (c), 4.5 Å (d), respectively. Purple and green in (c) and (d) represent gain and loss of electrons, respectively. Isovalue = 0.02. Differential charge densities of the GWG system with other distances are shown in Fig. S4.† | ||
Fig. 4 shows the HOMOs and LUMOs of nine typical configurations at 300 K to further understand the effects of different distances on the frontier molecular orbitals of the GWG systems. When the distances are larger than 6.0 Å, we found the HOMO or the LUMO is only from one of graphene layers. Interestingly, it is mainly from the graphene layer adsorbed by water molecules, which better explains why the density distribution curves of O atoms are not completely symmetrical in Fig. 2. The effect was better attributed to the fact that, water molecules would induce a little difference in the electronic structure near the two graphene layers. And the orbitals are from joint contribution of both upper layer and lower layer of graphenes, with HOMO-n, corresponding systems HOMO-2 (6.0 Å), HOMO-5 (6.5 Å), HOMO-23 (7.0 Å), HOMO-23 (8.0 Å), HOMO-6 (9.0 Å), respectively. These results indicate the adsorption of water could highly enhance the activity of electron orbitals of graphene. However, when the distances are less than 6.0 Å, the two layers of graphenes and the middle water molecules all contribute to both HOMOs and LUMOs. Especially, for d = 4.5 Å and 4.0 Å, it is good to show the overlap of the frontier orbitals between water and graphene layers.
 | ||
| Fig. 4 Typical orbital diagrams of HOMO and LUMO for different distances. As opposed to the features of those with distances less than 6.0 Å, we found that the contribution of HOMO or LUMO was only from one sheet of graphene bilayer when the distance reaches 6.0 Å, so we also marked the molecular orbital HOMO-n for including the other sheet of graphene bilayer. | ||
Additionally, we also calculated total density of states (DOS) and partial density of states (PDOS) of GWG system with the distances of 4.0, 4.5, 6.5, 9.0 Å, as well as orbital energies of GWG systems with different distances at 300 K, as shown in Fig. S6.† And we also present the contribution percentages of atomic orbitals to the HOMOs and the LUMOs of GWG systems with the different distances at 300 K in Table S1.† Combined with Fig. S6 and Table S1,† it can be seen that, for the relatively large interlayer distance, the effects of distance changes on the Fermi level are only at a scale of 0.01 eV, and the graphene exhibits semiconductor properties, which are hardly changed by water doping. Interestingly, unlike the cases of larger interlayer distances, the Fermi levels of 4.0 Å and 4.5 Å are clearly different since 2p orbital contributions of O atoms to the HOMOs and the LUMOs are significant changed.
The properties of the total, time-dependent, Mermin free energy (Etf) are important to address the stability of GWG, as shown in Fig. S7.† There, it can be seen that all systems reached dynamic equilibrium. Combined with Fig. 1 and 4, the results indicate that the relatively stable states of the GWG system are within the vicinity of d = 6.0 Å. Above d = 6.0 Å, there is a slight increase in the energy of GWG system, as the interlayer distance increases, because of the attraction between aqueous layer and graphene is weak. Below d = 6.0 Å, since water molecules are entropically frustrated when the distance reduces, which leads to Etf sharply increasing.
Conclusions
In summary, we have studied the effects of the interlayer distance on the water adsorption in-between two graphene layers, as well as the dynamic and electronic properties of a prototypical GWG system using the DFTB3 method. We show that the water molecules inside GWG resemble bulk water, with comparatively free motions and typical bulk-like dynamics, when the distance is large. The charge transfers are very small, at the scale of 0.001 e per water molecule. Meanwhile, the HOMO or the LUMO is only from one of the graphene layers. As the distance decreases, particularly at distances shorter than the vdW boundary, the sandwich structures of GWG reveal special properties, such as the ordering of the aqueous layer and the total increase of the Mermin free energy, however, the main HOMO and LUMO contributions arise from both graphene layers. Interestingly, when the distance is further reduced to 4.5 Å, OH bonds are almost parallel to bilayer graphene, oxygen atoms containing lone pair of electrons in water molecules are toward the graphene, leads to the electronic migration from the p orbitals of oxygen atoms to graphene, causing the aqueous layer to lose electrons. For d = 4.0 Å, the aqueous layer changes again drastically and becomes an electrons acceptor. This switch in behavior is explained in terms of the entropic frustration, causing a large number of OH bonds of non-hydrogen bonds to point towards the graphene bilayer, and leading to empty 1s orbitals of H atoms disturbed by delocalized π orbitals formed from the p electrons of C atoms.GWG sandwich structures like the one studied here have many potential applications, such as lubrication,36,58 regulation of water transport in graphene nano-channels,11 and the design of nanofluidic devices27 or coolant devices,59 etc. Our study points out that the different interlayer distances may induce interesting switching behaviors at an atomic level, affecting both geometric and electronic structures. Of special interest are cases with distances below the vdW boundary, where the interlayer GWG system shows unique electronic structures and anomalous conformations. Taken together, these results provide some theoretical foundation for further future studies, which might facilitate applications in lubrication, water transport, nanofludics and so on.
Acknowledgements
The work was supported by the National Basic Research Program of China (973 Program), the National Science Foundation of China (11374004 and 11374221) and Science and Technology Development Program of Jilin Province of China (20150519021JH). Z. W. acknowledges the Fok Ying Tung Education Foundation (142001) and High Performance Computing Center (HPCC) of Jilin University. A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection.Notes and references
- C. J. Shih, Q. H. Wang, S. C. Lin, K. C. Park, Z. Jin, M. S. Strano and D. Blankschtein, Phys. Rev. Lett., 2012, 109, 176101 CrossRef.
- A. Eftekhari-Bafrooei and E. Borguet, J. Am. Chem. Soc., 2010, 132, 3756–3761 CrossRef CAS PubMed.
- D. Argyris, N. R. Tummala, A. Striolo and D. R. Cole, J. Phys. Chem. C, 2008, 112, 13587–13599 CAS.
- L. D'urso, C. Satriano, G. Forte, G. Compagnini and O. Puglisi, Phys. Chem. Chem. Phys., 2012, 14, 14605–14610 RSC.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 11450–11458 CrossRef CAS.
- H. Li and X. C. Zeng, ACS Nano, 2012, 6, 2401–2409 CrossRef CAS PubMed.
- E. Voloshina, D. Usvyat, M. Schütz, Y. Dedkov and B. Paulus, Phys. Chem. Chem. Phys., 2011, 13, 12041–12047 RSC.
- V. Bianco and G. Franzese, Sci. Rep., 2014, 4, 4440 Search PubMed.
- G. Zilibotti, S. Corni and M. C. Righi, Phys. Rev. Lett., 2013, 111, 146101 CrossRef CAS.
- C. A. Palma, J. Bjork, M. Bonini, M. S. Dyer, A. Llanes-Pallas, D. Bonifazi, M. Persson and P. Samorı, J. Am. Chem. Soc., 2009, 131, 13062–13071 CrossRef CAS PubMed.
- W. Xiong, J. Z. Liu, M. Ma, Z. P. Xu, J. Sheridan and Q. S. Zheng, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 056329 CrossRef.
- J. Martí, J. Sala and E. Guàrdia, J. Mol. Liq., 2010, 153, 72–78 CrossRef PubMed.
- J. Y. Li, X. J. Gong, H. G. Lu, D. Li, H. P. Fang and R. H. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3687–3692 CrossRef CAS PubMed.
- I. Hamada, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195436 CrossRef.
- D. S. Yang and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4122–4126 CrossRef CAS PubMed.
- R. H. Zhou, X. Huang, C. J. Margulis and B. J. Berne, Science, 2004, 305, 1605–1609 CrossRef CAS PubMed.
- L. Hua, R. H. Zhou, D. Thirumalai and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16928–16933 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. Mcgovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- M. Choucair, P. Thordarson and J. A. Stride, Nat. Nanotechnol., 2009, 4, 30–33 CrossRef CAS PubMed.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. M. Wang, I. T. Mcgovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- X. Li, C. W. Magnuson, A. Venugopal, R. M. Tromp, J. B. Hannon, E. M. Vogel, L. Colombo and R. S. Ruoff, J. Am. Chem. Soc., 2011, 133, 2816–2819 CrossRef CAS PubMed.
- M. C. Gordillo and J. Martí, J. Phys. Chem. B, 2010, 114, 4583–4589 CrossRef CAS PubMed.
- T. A. Ho and A. Striolo, J. Chem. Phys., 2013, 138, 054117 CrossRef PubMed.
- F. Taherian, V. Marcon, N. F. A. Van Der Vegt and F. Leroy, Langmuir, 2013, 29, 1457–1465 CrossRef CAS PubMed.
- H. Ruuska and T. A. Pakkanen, Carbon, 2003, 41, 699–706 CrossRef CAS.
- J. S. Babu and S. P. Sathian, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 051205 CrossRef.
- Y. Qiao, X. Xu and H. Li, Appl. Phys. Lett., 2013, 103, 233106 CrossRef PubMed.
- I. Strauss, H. Chan and P. Král, J. Am. Chem. Soc., 2014, 136, 1170–1173 CrossRef CAS PubMed.
- G. Cicero, J. C. Grossman, E. Schwegler, F. Gygi and G. Galli, J. Am. Chem. Soc., 2008, 130, 1871–1878 CrossRef CAS PubMed.
- V. Tozzini, D. K. Chaturvedi and M. P. Tosi, Phys. B, 1997, 240, 92–97 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- R. Quhe, J. H. Ma, Z. S. Zeng, K. Tang, J. X. Zheng, Y. Y. Wang, Z. Y. Ni, L. Wang, Z. X. Gao, J. J. Shi and J. Lu, Sci. Rep., 2013, 3, 1794 Search PubMed.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951–954 CrossRef CAS PubMed.
- A. Pertsin and M. Grunze, J. Phys. Chem. B, 2004, 108, 1357–1364 CrossRef CAS.
- B. K. Yen, B. Schwickert and M. F. Toney, Appl. Phys. Lett., 2004, 84, 4702–4704 CrossRef CAS PubMed.
- S. Wangmo, R. X. Song, L. Wang, W. W. Jin, D. J. Ding, Z. G. Wang and R. Q. Zhang, J. Mater. Chem., 2012, 22, 23380 RSC.
- Y. Meng, Q. Wu, L. Chen, S. Wangmo, Y. Gao, Z. G. Wang, R. Q. Zhang, D. J. Ding, T. A. Niehaus and T. Frauenheim, Nanoscale, 2013, 5, 12178–12184 RSC.
- P. Goyal, M. Elstner and Q. Cui, J. Phys. Chem. B, 2011, 115, 6790–6805 CrossRef CAS PubMed.
- R. X. Song, S. Wangmo, M. S. Xin, Y. Meng, P. Huai, Z. G. Wang and R. Q. Zhang, Nanoscale, 2013, 5, 6767–6772 RSC.
- Z. T. Li, Y. J. Wang, A. Kozbial, G. Shenoy, F. Zhou, R. Mcginley, P. Ireland, B. Morganstein, A. Kunkel, S. P. Surwade, L. Li and H. T. Liu, Nat. Mater., 2013, 12, 925–931 CrossRef CAS PubMed.
- M. Elstner, Th. Frauenheim and S. Suhai, J. Mol. Struct.: THEOCHEM, 2003, 632, 29–41 CrossRef CAS.
- Y. Yang, H. B. Yu, D. York, Q. Cui and M. Elstner, J. Phys. Chem. A, 2007, 111, 10861–10873 CrossRef CAS PubMed.
- M. Gaus, Q. Cui and M. Elstner, J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS PubMed.
- N. Choudhury and B. M. Pettitt, J. Phys. Chem. B, 2005, 109, 6422–6429 CrossRef CAS PubMed.
- H. F. Ye, H. W. Zhang, Z. Q. Zhang and Y. G. Zheng, J. Adhes. Sci. Technol., 2012, 26, 1897–1908 CAS.
- W. C. Swope, H. C. Andersen, P. H. Berens and K. R. Wilson, J. Chem. Phys., 1982, 76, 637 CrossRef CAS PubMed.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS PubMed.
- J. Y. Li, T. Liu, X. Li, L. Ye, H. J. Chen, H. P. Fang, Z. H. Wu and R. H. Zhou, J. Phys. Chem. B, 2005, 109, 13639–13648 CrossRef CAS PubMed.
- X. Wu and X. Lu, J. Am. Chem. Soc., 2007, 129, 2171–2177 CrossRef CAS PubMed.
- X. Dai, C. Cheng, W. Zhang, M. S. Xin, P. Huai, R. Q. Zhang and Z. G. Wang, Sci. Rep., 2013, 3, 1341 Search PubMed.
- M. Elstner, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- S. Nénon, R. Méreau, S. Salman, F. Castet, T. V. Regemorter, S. Clima, D. Beljonne and J. Cornil, J. Phys. Chem. Lett., 2012, 3, 58–63 CrossRef.
- Y. Yang, H. Yu, D. York, M. Elstner and Q. Cui, J. Chem. Theory Comput., 2008, 4, 2067–2084 CrossRef CAS PubMed.
- M. P. Lourenço, L. Guimarães, M. C. Da Silva, C. de Oliveira, T. Heine and H. A. Duarte, J. Phys. Chem. C, 2014, 118, 5945–5953 Search PubMed.
- J. C. Slater, J. Chem. Phys., 1964, 41, 3199 CrossRef CAS PubMed.
- Y. C. Wang, H. Y. Liu, J. Lv, L. Zhu, H. Wang and Y. M. Ma, Nat. Commun., 2011, 2, 563 CrossRef PubMed.
- A. Ambrosetti, F. Ancilotto and P. L. Silvestrelli, J. Phys. Chem. C, 2013, 117, 321–325 CAS.
- T. T. Baby and S. Ramaprabhu, J. Appl. Phys., 2010, 108, 124308 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13736a |
| This journal is © The Royal Society of Chemistry 2015 |