
Acid base co-crystal converted into porous carbon material for energy storage devices†
Abstract
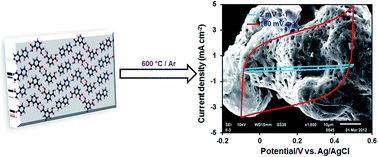
A simple and facile method is adopted for the synthesis of pure and catalyst free carbon material for supercapacitor applications. In a co-crystal synthesis, the precursors (isophthalic acid and a base, 4,4′-bipyridine) are arranged in regular pattern, followed by carbonization at 600 °C under an inert atmosphere to produce pure carbon material, CIN-600. The obtained sample is characterized by many techniques, such as powder X-ray diffraction (PXRD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDS), and gas adsorption analysis. The gas adsorption and microscopic analysis demonstrated the high porosity of the carbon sample and its irregular geometry. Owing to the excellent porosity and electrical conducting properties, CIN-600 showed enhanced capacitive performance when used as an electrode material in electric double layer capacitors. The specific capacitance of the sample was ca.181.3 F g−1 at 2 mV s−1 and maintained 91.3% of its initial capacitance in a long-term cycling test.
 Please wait while we load your content...
Please wait while we load your content...

