
Supercapacitor devices for energy storage and capacitive dye removal from aqueous solutions†
Kaiyuan Shi and
Igor Zhitomirsky*
Department of Materials Science and Engineering, McMaster University, 1280 Main Street West Hamilton, ON, Canada L8S 4L7. E-mail: zhitom@mcmaster.ca; Tel: +1-905-525-9140
First published on 24th November 2014
Abstract
Multiwalled carbon nanotubes (MWCNT) are coated with nitrogen doped activated carbon (NC) for applications in electrochemical supercapacitor electrodes and devices. The capacitive behavior of the electrodes and devices is tested in solutions of organic dyes. The process involves energy accumulation in electrical double layers. The accumulated energy may be further utilized to remove dyes with a similar scheme via discharge of the supercapacitor, also fully regenerating the electrode materials for further use. Proof of concept investigations involve the testing of anionic and cationic dyes and analysis of the influence of dye concentration, charge as well as charge to mass ratio on the capacitance, impedance, specific power and energy of the devices. Cyclic voltammetry and chronopotentiometry data, obtained in different voltage windows, are used for the optimization of device performance. The discussion of quartz crystal microbalance data provides an insight into the factors which govern the charge–discharge behavior. The devices show good cyclic stability. This method offers the advantage of saving energy and applicability to a variety of different dyes.
1. Introduction
Electrochemical supercapacitors (ES) are currently under development for energy storage systems due to their high power density, fast charge/discharge and good cycling stability.1–4 There are many excellent reviews describing new electrode materials, energy storage mechanisms, electrolytes, and the design and performance of ES.5–10 Efficient charge storage can be achieved in electrochemical double layers of high surface area carbon materials.5,11–14 The charge storage in pseudocapacitive materials is based on reversible electrochemical reactions.5,15–17 Significant interest has been generated in the development of asymmetric capacitors, based on two different electrodes, and hybrid devices, containing capacitive and battery electrodes.6,18 Many fundamental investigations were focused on the synthesis of advanced electrode materials with high capacitance, small particle size and high surface area.5–9 Recent studies19–21 highlighted the importance of the fabrication of porous ES electrodes with high active mass loading and high active material to current collector mass ratio. High power ES have promising applications in electronic devices, hybrid and electric vehicles.A new wave of interest in the ES technology is related to the application of ES devices for capacitive deionization (CDI)22,23 which was found to be a robust, energy efficient, and cost effective technology for desalination of water with a low or moderate salt content. In the CDI technology, the charging process results in removal of ions from the solutions and their accumulation in electrodes for energy storage in electrical double layers. The energy, released during the discharge process, can be used for the charging of neighbouring cells. Therefore, the CDI technology offers the advantages of energy saving.22 Ongoing research is focused on the optimization of operation modes, investigation of the kinetics of ion removal and development of efficient electrode materials.23–26 Special attention is focused on the surface modification of electrode materials. It was shown that the fabrication of porous carbon electrodes with high surface area is of critical importance for further development of the CDI technology. Other important properties of electrode materials are: high chemical stability, high electronic conductivity, surface charge and good wetting behaviour.22
Recent advances in the CDI technology indicate that ES are promising devices for the capacitive removal of other charged species from aqueous solutions. Of special interest is the possibility of capacitive dye removal from the solutions. Organic dyes are widely used for many applications, such as solar cells,27 photocatalysis,28 transistors,29 biological and chemical sensors,30 lasers,31 medicine,32 textile industry33 and high tech applications.33 The increased utilization of organic dyes for various applications given their rich diversity and complexity mandates the development of robust methods for dye removal from the chemical waste. Physical adsorption, chemical and biochemical treatment are important methods for dye removal.34–36 Significant progress has also been made in the development of chemical oxidation and reduction, photocatalytic and ozonation methods.37 Electrochemistry is widely recognized as an essential tool for dye removal from wastewaters.38,39 Different electrochemical techniques have been developed, such as electrocoagulation, electrochemical reduction, electrochemical oxidation, photoassisted electrochemical methods and other techniques.39,40 The methods have some inherent problems,39 such as significant energy consumption, electrode degradation and limited range of applications. To date, no single method has demonstrated the combined facets of wide chemical compatibility, low energy usage, inexpensive processing, and high recyclability.39 The diversity and complexity of the dyes generates a need in new methods for dye removal.
In a previous investigation,41 we observed significant changes of the mass of quartz crystal microbalance resonators in response to an applied electric field in the dilute solutions of the anionic methyl blue dye. Such mass gain of the electrodes resulted from the accumulation of the dye molecules at the electrode surface. It was suggested that the accumulation of the charged dyes at the electrode can be used for energy storage and capacitive dye removal. However, previous investigations were limited to the quartz crystal microbalance studies of mass gain in the dilute solutions of the anionic methyl blue dye. No direct observation of capacitive behaviour and energy storage was reported.
There is a need in the proof-of-concept studies, involving demonstration of capacitive properties and energy storage in individual electrodes and two electrode devices in dye solutions, including solutions of different cationic and anionic dyes at different concentrations. The important task is the measurements of capacitance of individual electrodes and devices using cycling voltammetry and constant current charge–discharge techniques. Another important task is the measurements and analysis of impedance and AC capacitance, derived from the impedance data. Such studies must be performed in more concentrated solutions in order to achieve sufficient energy accumulation in the electrical double layers. Critical to the development and testing of robust ES cells is the utilization of efficient electrodes with high active mass loadings. In contrast to previous investigations, the microbalance data must be obtained at different voltages. One of the important parameters of the supercapacitor devices is the voltage window, which must be optimized using electrochemical testing and quartz crystal microbalance data. The comparison of the data for different dyes with different structures and charges can provide valuable information, related to the feasibility of energy storage in devices, containing organic dyes as electrolytes. It is important to analyze the Coulombic efficiency and power-energy characteristics of the devices. Such proof-of concepts studies were the goal of our work, focused on the analysis of capacitive behavior of electrodes and devices in solutions of different dyes.
In addition to addressing the proof-of-concept questions, we analyzed the data for different dyes, which provide an insight into the influence of dye structure and charge on capacitive behavior of the devices. Moreover, the results presented below indicated that the method of capacitive dye removal can be used for energy storage. Electrode regeneration resulted in energy release that may be further utilized for continued purification via charging of neighboring cells or other applications. This method offers the advantage of vastly improved energy utilization. The dyes can be reused after the electrode regeneration. Our results demonstrate the wide applicability of capacitive dye removal that can be used for both cationic and anionic dyes.
2. Experimental procedures
2.1 Materials
Safranin (SF), calcon (CL), amaranth (AM), N-methylpyrrolidone, polyvinylidene fluoride (PVDF), KOH, polyvinyl butyral (PVB) (Sigma-Aldrich), MWCNT (Arkema), Ni foam (95% porosity, Vale) were used. For the fabrication of NC–MWCNT, the PPy coated MWCNT, prepared according to our previous work,42 were mixed with KOH and then carbonized and activated in a nitrogen gas atmosphere at 700 °C for 2 h.2.2 Quartz crystal microbalance studies
Quartz crystal microbalance (Princeton Applied Research, QCM 922) was used (Fig. 1) for the in situ analysis of capacitive adsorption and desorption of dyes. The electrochemical cell included a 9 MHz quartz resonator, containing Au electrodes, coated with NC–MWCNT, and a Pt counter electrode. The NC–MWCNT coating was obtained by casting of 2 wt% NC–MWCNT suspension in N-methylpyrrolidone solvent, containing 0.1 wt% PVDF binder. The mass variations of the quartz resonators were calculated using the Sauerbrey's equation.43,44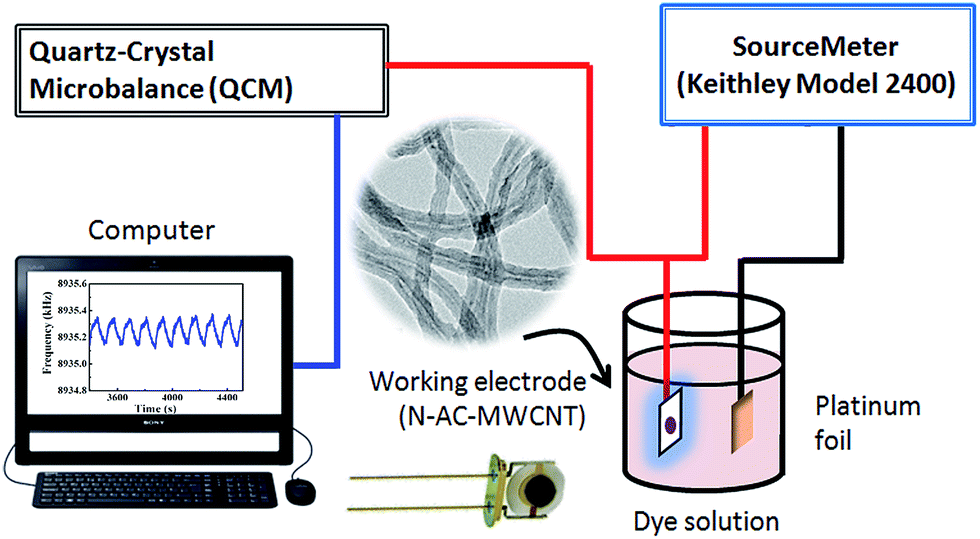 | ||
| Fig. 1 Schematic of the QCM analysis system for capacitive dye removal. | ||
The QCM studies of capacitive adsorption and desorption were performed in the pulse mode, using positive and negative pulses with a constant voltage of 1 V or by applying a series of positive or negative pulses of increasing amplitude in the range of 0.6–1.4 V. The electrode separation distance was 1 cm. The ON and OFF times were 1 min. A Keithley (model 2400) power supply was used for the QCM studies.
2.3 Fabrication of electrodes and cells for electrochemical testing
Electrodes for cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) studies were prepared by casting of suspension, containing NC–MWCNT and 5% PVDF binder in N-methylpyrrolidone, on stainless steel foil, which was then dried in oven. The mass of the dried material casted on the stainless steel was 1 mg cm−2. The electrodes for ES devices were prepared by impregnation of Ni foam current collectors using suspensions of NC–MWCNT in ethanol, containing 5% PVB binder. The ES cells contained two electrodes with mass loading of 15 mg cm−2, separated by a porous polymer membrane, and 400 mg L−1 dye aqueous solution as an electrolyte.2.4 Characterization methods
Electron microscopy investigations were performed using Osiris field emission transmission electron microscope (TEM) and JEOL JSM-7000F scanning electron microscope (SEM). A potentiostat (Princeton Applied Research, PARSTAT 2273) was used for electrochemical studies. A three electrode cell contained a working electrode with area 1 cm2, a Pt gauze counter electrode and a saturated calomel electrode (SCE) as a reference electrode. Cyclic voltammetry (CV) studies were performed using 100, 200 and 400 mg L−1 dye solutions within a potential range of −1 to 0 V (or 0 to +1 V) versus SCE at scan rates of 0.5–20 mV s−1. The CV data was used for the calculation of capacitance of electrodes Cm = Q/mΔV, where Q is the charge, m is electrode mass and ΔV is the potential window. The complex impedance Z* = Z′ − iZ′′ was measured at the amplitude of the alternating current (AC) signal of 5 mV at frequencies of 0.01 Hz to 100 kHz. The galvanostatic charge–discharge behaviour of the ES cells was investigated at different current densities in the range of 0.5–10 mA cm−2 using a battery analyzer (MTI corporation, BST8). The gravimetric Ccm = Cc/2m and areal Ccs = Cc/S capacitances of the coin cells were calculated from the discharge data, where Cc is the cell capacitance, m is mass of one electrode and S is electrode area.3. Results and discussion
Fig. 2 shows solutions and chemical structures of the dyes used in this investigation. We selected cationic SF, anionic CL and AM dyes that have many important applications. SF and its derivatives are used in the metal electroplating industry,45 medicine46 and photovoltaic devices.47 CL and AM belong to a large family of azo dyes, which have many applications in the textile and pharmaceutical industry,48–50 and mineral flotation.51 Fig. 2 indicates that structures of SF and CL have one cationic or anionic charged group, respectively. The structure of AM is similar to that of CL. However, AM has three anionic groups. In this investigation, the comparison of the experimental data for AM and CL provided an insight into the influence of charge of the molecules on the capacitive behaviour. The molecular volumes, areas, hydrophobicities and other data for the dyes, obtained by computation methods, are described in the ESI (Table S1†). | ||
| Fig. 2 (A) Aqueous solutions of (a) SF, (b) CL, (c) AM and chemical structures of (B) SF, (C) CL and (D) AM. | ||
The use of NC–MWCNT as an electrode material for capacitive dye removal offered the advantages of high surface area of NC and high conductivity of MWCNT. Fig. 3A and B presents typical TEM images of NC–MWCNT at different magnifications. The TEM image at lower magnification (Fig. 3A) showed uniform diameter of NC–MWCNT. The higher magnification image (Fig. 3B) indicated the formation of relatively uniform NC coatings. The NC–MWCNT material has a surface area of 1889 m2 g−1 (Fig. S1† and related data analysis in the ESI†).
 | ||
| Fig. 3 (A) TEM and (B) HRTEM images of NC–MWCNT, arrows show NC layer. | ||
Fig. 4(A and B) shows CVs for NC–MWCNT electrodes in SF and CL solutions. The area of the CVs increased with increasing dye concentration, indicating the increase in number of adsorbed dye molecules, contributing to the formation of the electrical double layer and charge–discharge currents. The capacitances, calculated from the CV data are presented in Fig. 4C. The electrodes showed comparable capacitances of 39 and 41 F g−1 in the solutions of SF and CL dyes, respectively, at dye concentration of 100 mg L−1. However, the capacitance achieved in SF solutions at concentrations of 200 and 400 mg L−1 was higher than the capacitance in CL solutions of similar concentrations. The capacitances in 400 mg L−1 solutions of SF and CL dyes were found to be 145 and 103 F g−1, respectively.
 | ||
| Fig. 4 (A and B) CVs at a scan rate of 0.5 mV s−1 for NC–MWCNT electrodes in the dye solutions of (A) SF and (B) CL with concentrations of (a) 100, (b) 200 and (c) 400 mg L−1, (C) specific capacitances, calculated from the CV data, (D) Nyquist plot of complex impedance Z* = Z′ − Z′′ for different solutions: (a) 100, (b) 200 and (c) 400 mg L−1 SF and (d) 100, (e) 200 and (f) 400 mg L−1 CL. | ||
Fundamental studies performed by other groups showed that capacitance of porous carbon materials depends on the size of the electrolyte molecules and pore size of carbon.52–56 They found53 that using ionic liquid electrolytes and activated carbons with controlled pore size, the specific capacitance of 160 F g−1 can be achieved, which is much higher than the specific capacitance reported (50–100 F g−1) for commercial activated carbons. The size of SF and CL dyes (Table S1†) is comparable with the size of the ionic liquid molecules.53 The specific capacitance of NC–MWCNT electrodes, achieved in the SF solutions was comparable with specific capacitance of the carbon materials in ionic liquid electrolytes. It is important to note that the solutions of SF and CL dyes contained large cation or anion, respectively, and smaller counter ions. In this case, the capacitive performance is strongly influenced by large ions. The lower capacitance, achieved in the CL solutions, can be partially attributed to larger mass and size of the CL molecules (Table S1†), which can result in lower ion mobility. It is known54 that charge storage in double layers is influenced by ion transport. The difference in performance of NC–MWCNT electrodes in the solutions of SF and CL dyes can also be attributed to electrostatic interactions of NC–MWCNT and SF+ cations or CL− anions. Recent studies have shown that cationic and anionic transports in pores depend57–60 on point of zero charge of the electrode material. It was demonstrated that negatively charged carbon nanotubes promoted cationic transport.58 The potential of zero charge was found to be different from the potential of zero mass change,61 that separates the mass change of the electrode due to adsorption of cations and anions. Other investigations emphasized the influence of hydrophobic interactions with pore walls.62
The investigations of the electrochemical impedance Z* = Z′ − iZ′′ of the NC–MWCNT electrodes in SF and CL solutions, presented in the Nyquist plot (Fig. 4D), showed that the increase in dye concentration resulted in decreasing resistance R = Z′. The decrease in Z′′ with increasing dye concentration indicated increasing capacitance C′ = Z′′/ω|Z|2.
The capacitive dye removal has also been investigated using a QCM method, which is an efficient tool for monitoring the incorporation of ions into the ES electrodes during charge and discharge cycles.61,63,64 Testing results showed that the application of negative or positive pulses to the NC–MWCNT electrodes in SF or CL solutions, respectively, resulted in mass gain, attributed to incorporation of cationic SF or anionic CL species into the electrode (Fig. 5A and B). The mass gain versus time curves for the electrodes are typically characterized by several domains, representing complex interactions of ions and porous electrodes.61 The electrode mass decreased during the OFF time. The experiments showed periodic variations in the electrode mass. Such periodic variations, related to dye removal, indicate a possibility of regeneration of the electrode material. The increase in pulse amplitude resulted in increasing mass gain (Fig. 5B and D and Table S2†), indicating the increase in amount of ions incorporated into the electrodes.
 | ||
| Fig. 5 QCM data for NC–MWCNT coated resonators in 50 mg L−1 solutions of (A and B) SF and (C and D) CL, tested using (A) negative and (C) positive pulses with a constant voltage of 1 V or by applying a series of (B) negative or (D) positive pulses of increasing amplitude in the range of 0.6–1.4 V. | ||
The results of CV and QCM testing paved the way for the fabrication of ES devices that enabled the removal of dyes from aqueous solutions and energy storage in electrical double layers of NC–MWCNT electrodes (Fig. S2†). The charge–discharge behavior of the two-electrode devices was analyzed in different voltage windows (Fig. 6A–F). Fig. 6(A and D) shows CVs in different voltage windows for the ES devices, containing SF or CL electrolytes at a scan rate of 2 mV s−1. The increase in the voltage window above 0.9 V resulted in significant increase in current, attributed to redox reactions. The galvanostatic charge–discharge curves obtained at a current density of 3 mA cm−2 showed significant voltage drop at the beginning of the discharge for voltage windows above 0.9 V (Fig. 6(B and E)). Such voltage drop V = IR (I – current) can be attributed to increased electrical resistance R of the cells. The cell capacitances and Coulombic efficiencies decreased in the potential windows above 0.9–1 V (Fig. 6C and F). Therefore, further investigations were performed in the potential window of 0.9 V. Fig. 7(A and B) shows CVs at different scan rates in the voltage window of 0.9 V. The larger area of the CVs obtained for the cells, containing SF electrolyte (Fig. 7A), compared to the cells, containing CL electrolyte (Fig. 7B), indicated higher capacitance of the SF cells. The capacitance, calculated from the CV data is plotted versus scan rate in the Fig. 7C. The cells, containing SF electrolyte, showed higher capacitance, compared to the cells containing CL electrolyte. The difference was especially evident at scan rates above 2 mV s−1. The lower capacitance of the cells, containing CL electrolyte, can result from higher resistance of such cells (Fig. S3†). Moreover the analysis of the impedance data showed that the real component C′ = Z′′/ω|Z|2 of complex capacitance for CL containing cells was lower, compared to that of SF containing cells (Fig. S3†). This result correlated with capacitance data, calculated from CVs for the same cells.
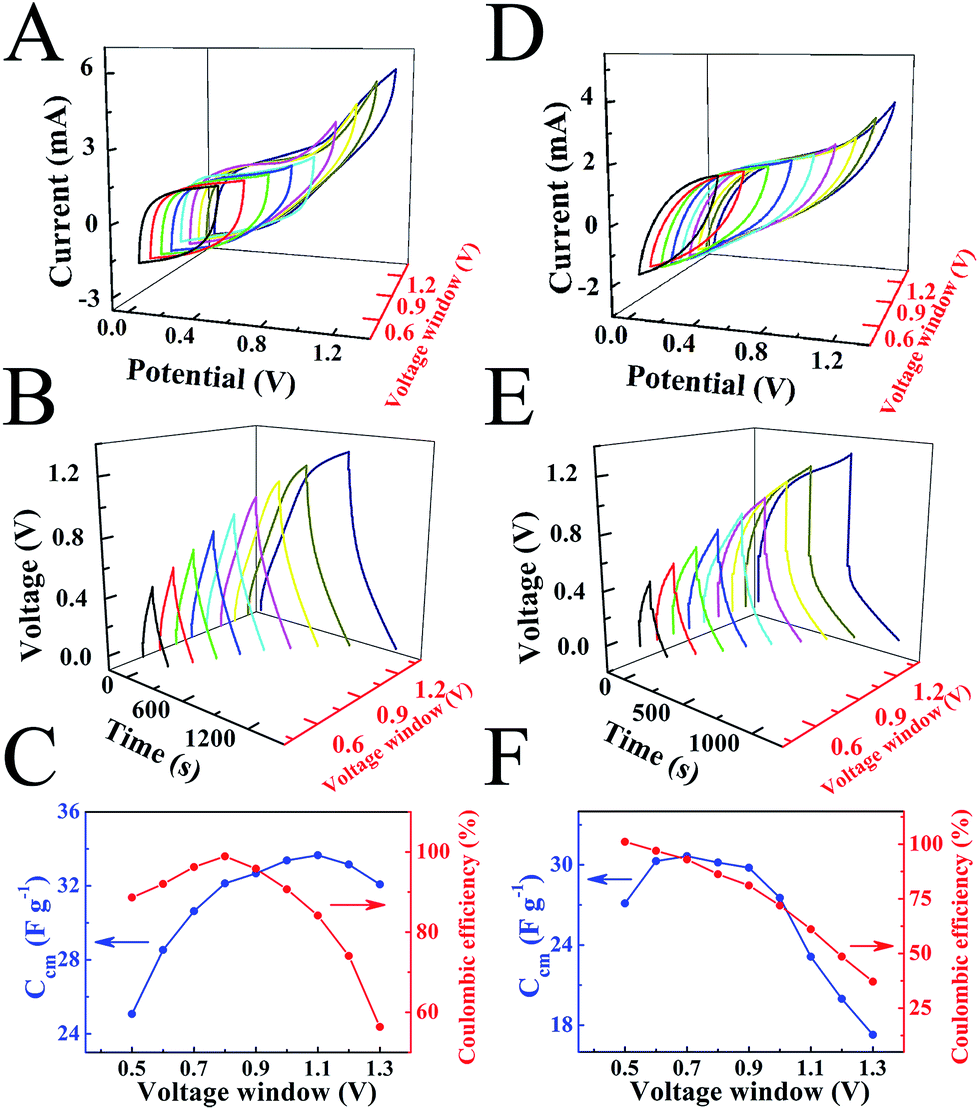 | ||
| Fig. 6 Capacitive behavior in different potential windows (from 0.5 to 1.3 V) for ES cells, containing two NC–MWCNT electrodes in 400 mg L−1 (A–C) SF and (D–F) CL solutions as electrolytes. (A and D) CVs at a scan rate of 2 mV s−1, (B and E) charge–discharge behavior at a current density of 3 mA cm−2, (C and F) Ccm calculated from discharge curves and Columbic efficiency versus the width of the potential window. The mass loadings of individual electrodes was 15 mg cm−2. | ||
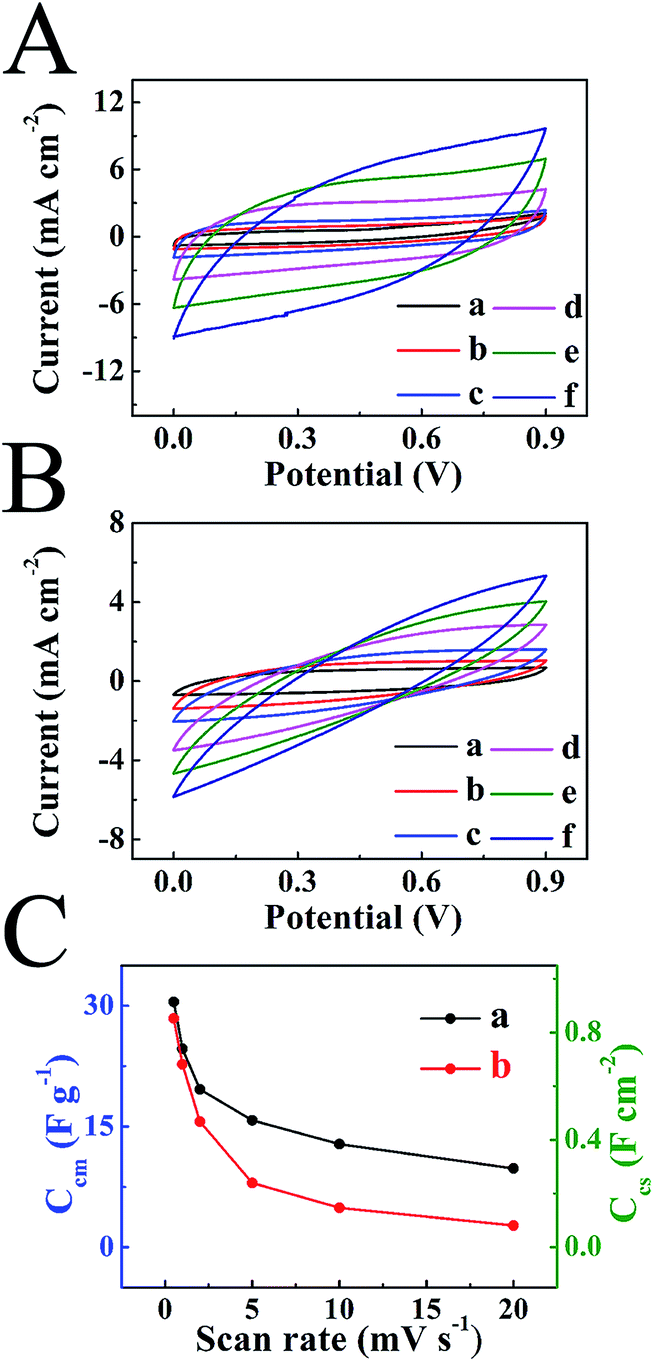 | ||
| Fig. 7 CVs in a voltage window of 0.9 V for ES cells prepared using 400 mg L−1 (A) SF and (B) CL electrolytes at scan rates of (a) 0.5, (b) 1, (c) 2, (d) 5, (e) 10 and (f) 20 mV s−1. (C) Ccm and Ccs calculated from CVs versus scan rate for (a) SF and (b) CL electrolytes. The mass loadings of individual electrodes of ES cells is 15 mg cm−2. | ||
Fig. 8(A and B) shows charge–discharge behavior of ES devices, containing two NC–MWCNT electrodes and SF or CL electrolytes in a voltage window of 0.9 V. The charge–discharge curves showed nearly symmetrical triangular shape, however voltage drop was observed at the beginning of the discharge curve. This voltage drop U = IR (I – current, R – resistance) was especially evident for the cells, containing CL electrolyte, due to higher resistance. The devices, based on SF electrolyte showed higher capacitance and better capacitance retention (Fig. 8C) in the current density range of 1–8 mA cm−2. The energy densities and power densities for the devices were presented in Fig. 8D. The devices (Fig. 8D, insets), based on SF and CL showed maximum energy density of 5.3 and 4.1 mW h g−1 and power density of 525 and 137 mW g−1, respectively.
 | ||
| Fig. 8 (A and B) Charge–discharge behavior of ES cells in (A) SF and (B) CL electrolytes at current densities of (a) 2, (b) 3, (c) 4, (d) 6 and (e) 8 mA cm−2; (C) Ccm and Ccs calculated from the discharge data versus current density and (D) Ragone plot for ES cells (insets) containing 400 mg L−1 (a) SF and (b) CL solutions as electrolytes. The NC–MWCNT mass loadings of individual electrodes of ES cells is 15 mg cm−2. | ||
The SF and CL dyes contain only one charged group per molecule. We suggest that capacitive removal is especially attractive for the dyes with several charged groups per molecule and higher charge to mass ratio. As pointed out above, the structure of AM dye is similar to that of CL dye, however, the AM molecule has three charged groups. The higher charge to mass ratio of AM molecules can be beneficial for their electromigration during charge and discharge. The ES cells, containing AM electrolyte, showed improved capacitive behavior, compared to the cells, containing CL electrolyte of the same concentration. The improved capacitive behavior is indicated by the larger area of CVs and nearly triangular shape of charge–discharge curves with reduced V = IR drop at the beginning of discharge in the voltage window of 0.9 V (Fig. 9A and B). The CVs obtained in larger voltage windows showed significant deviation from the box shape and increasing current related to electrochemical decomposition of water (Fig. S4A†). The charge–discharge curves showed the increase in voltage drop with increasing voltage window (Fig. S4B†). Therefore, similar to SF and CL cells, the voltage window of the AM cells was optimized at 0.9 V. Fig. 9C and D shows capacitances, calculated from CVs (Fig. 9A) and galvanostatic discharge (Fig. 9B) data. The AM cells showed higher capacitance and improved capacitance retention at high charge discharge rates, compared to CL cells. The capacitance of the AM cell at a scan rate of 20 V s−1 was 9.0 F g−1 (0.27 F cm−2), whereas the capacitance of the CL cell at the same scan rate was only 2.7 F g−1 (0.08 F cm−2). The capacitance of the AM cell at a discharge current of 8 mA cm−2 was 19.5 F g−1 (0.60 F cm−2), however the capacitance of CL cell at the same discharge current was only 9.60 F g−1 (0.29 F cm−2). The difference can result from higher mobility of the AM dye, which has higher charge and higher charge to mass ratio, compared to CL. The analysis of the impedance data for the cells, containing AM electrolyte showed lower resistance R = Z′ of the AM cells (Fig. S5A†), compared to the resistance of the CL cells. The lower resistance can provide improved power density. Moreover the AM cells showed higher AC capacitance C′ (Fig. S5B†), compared to the capacitance of CL cells. The capacitance of the AM cell, calculated from the discharge data increased with increasing voltage window and showed a maximum at a voltage of 1.1 V (Fig. 9E). The Coulombic efficiency (Fig. 9E) decreased with increasing voltage window, especially at voltages above 0.9 V. Fig. 9F presents a Ragone plot for the cell, containing AM electrolyte. The cell showed maximum energy density of 7.0 mW h g−1 and maximum power density of 337 mW g−1. The higher power density of the cell, containing AM electrolyte, compared to the cell containing CL electrolyte are attributed to higher capacitance and lower resistance of the AM cell.
 | ||
| Fig. 9 (A) CVs at scan rates of (a) 0.5, (b) 1, (c) 2, (d) 5, (e) 10 and (f) 20 mV s−1 and (B) charge–discharge curves at current densities of (a) 2, (b) 3, (c) 4, (d) 6 and (e) 8 mA cm−2 in a voltage window of 0.9 V, (C) Ccm and Ccs, calculated from the CV data versus scan rate, (D) Ccm and Ccs, calculated from the discharge data versus current density, (E) Coulombic efficiency and capacitance, calculated from the discharge data versus the width of the voltage window and (F) Ragone plot for ES cells, containing two NC–MWCNT electrodes in 400 mg L−1 AM electrolyte. | ||
The use of capacitive dye removal offers the advantage of efficient energy utilization, because the dyes, removed from solutions are used for energy storage in the electrical double layers. On the discharge cycle, this energy may be utilized to charge an analogous ES for dye removal or other applications. Turning again to the QCM data it should be mentioned that the method allows electrode regeneration. The dyes released during the discharge cycle can be used for other applications. This suggestion was supported by the analysis of cyclic stability of the cells. The AM cells showed 85.3 and 80.4% capacitance retention after 1000 and 3000 cycles, respectively (Fig. S6†). Quartz crystal microbalance studies of efficiency of dye removal (Fig. S7†) showed that one charging cycle resulted in the removal of 14.65% of AM dye from 50 mg L−1 AM dye solution at a deposition voltage of 1 V. The mass ratio of removed dye material to the mass of electrode material was 80 mg g−1. It is expected that further advances in the capacitive dye removal method can be achieved by the development of efficient electrodes with high porosity and this method will be used for other cationic and anionic dyes.
4. Conclusions
NC–MWCNT electrodes with high surface area have been demonstrated for capacitive removal of cationic SF, anionic CL and AM dyes. These electrodes offer a possibility of building ES devices that simultaneously remove dyes and store energy in an electrical double layer. This energy may be further utilized to remove dyes with a similar scheme via discharge of the ES, also fully regenerating the electrode materials for further use. QCM data showed reversibility of the dye accumulation and removal from the electrodes. ES devices based on the solutions of anionic and cationic dyes as electrolytes showed promising capacitive behaviour and good cycling stability. The devices, based on anionic AM dye showed improved capacitive behaviour, compared to electrodes with anionic CL dye with similar structure due to higher charge of the AM dye. The method offers the advantages of energy saving, reuse of dyes and electrodes and can be utilized for the removal of various cationic and anionic dyes.Acknowledgements
The authors acknowledge NSERC Canada for the funding and Vale Company for Ni foam.References
- Z. Yu, C. Li, D. Abbitt and J. Thomas, J. Mater. Chem. A, 2014, 2, 10923–10929 CAS.
- Z. Yu, B. Duong, D. Abbitt and J. Thomas, Adv. Mater., 2013, 25, 3302–3306 CrossRef CAS PubMed.
- Y. Wang, R. Yang, Y. Wei, Z. Zhao and M. Li, RSC Adv., 2014, 4, 45318–45324 RSC.
- M. Zhi, S. Liu, Z. Hong and N. Wu, RSC Adv., 2014, 4, 43619–43623 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. Yan, Q. Wang, T. Wei and Z. Fan, Adv. Energy Mater., 2014, 4, 1300816 Search PubMed.
- R. KÖtz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS PubMed.
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef CAS PubMed.
- X. Zhang, H. Zhang, C. Li, K. Wang, X. Sun and Y. Ma, RSC Adv., 2014, 4, 45862–45884 RSC.
- B. An, S. Xu, L. Li, J. Tao, F. Huang and X. Geng, J. Mater. Chem. A, 2013, 1, 7222–7228 CAS.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880–7903 CrossRef CAS PubMed.
- S. Boukhalfa, D. Gordon, L. He, Y. B. Melnichenko, N. Nitta, A. Magasinski and G. Yushin, ACS Nano, 2014, 8, 2495–2503 CrossRef CAS PubMed.
- L. Qie, W. Chen, H. Xu, X. Xiong, Y. Jiang, F. Zou, X. Hu, Y. Xin, Z. Zhang and Y. Huang, Energy Environ. Sci., 2013, 6, 2497–2504 Search PubMed.
- T. Brousse, M. Toupin and D. Belanger, J. Electrochem. Soc., 2004, 151, A614–A622 CrossRef CAS PubMed.
- D. Guo, X. Yu, W. Shi, Y. Luo, Q. Li and T. Wang, J. Mater. Chem. A, 2014, 2, 8833–8838 CAS.
- C.-W. Lee, S.-B. Yoon, S.-M. Bak, J. Han, K. C. Roh and K.-B. Kim, J. Mater. Chem. A, 2014, 2, 3641–3647 CAS.
- F. Wang, S. Xiao, Y. Hou, C. Hu, L. Liu and Y. Wu, RSC Adv., 2013, 3, 13059–13084 RSC.
- M. Sevilla, W. Gu, C. Falco, M. M. Titirici, A. B. Fuertes and G. Yushin, J. Power Sources, 2014, 267, 26–32 CrossRef CAS PubMed.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917–918 CrossRef CAS PubMed.
- Y. Munaiah, B. G. Sundara Raj, T. Prem Kumar and P. Ragupathy, J. Mater. Chem. A, 2013, 1, 4300–4306 CAS.
- S. Porada, R. Zhao, A. van der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS PubMed.
- S. Porada, D. Weingarth, H. V. M. Hamelers, M. Bryjak, V. Presser and P. M. Biesheuvel, J. Mater. Chem. A, 2014, 2, 9313–9321 CAS.
- S.-I. Jeon, J.-G. Yeo, S. Yang, J. Choi and D. K. Kim, J. Mater. Chem. A, 2014, 2, 6378–6383 CAS.
- H. Wang, L. Shi, T. Yan, J. Zhang, Q. Zhong and D. Zhang, J. Mater. Chem. A, 2014, 2, 4739–4750 CAS.
- H. Li, F. Zaviska, S. Liang, J. Li, L. He and H. Y. Yang, J. Mater. Chem. A, 2014, 2, 3484–3491 CAS.
- P. Koli, RSC Adv., 2014, 4, 46194–46202 RSC.
- R. Abe, K. Shinmei, N. Koumura, K. Hara and B. Ohtani, J. Am. Chem. Soc., 2013, 135, 16872–16884 CrossRef CAS PubMed.
- M. Gsanger, E. Kirchner, M. Stolte, C. Burschka, V. Stepanenko, J. Pflaum and F. Wurthner, J. Am. Chem. Soc., 2014, 136, 2351–2363 CrossRef PubMed.
- D. Geiler, S. Stufler, H.-G. Lohmannsroben and N. Hildebrandt, J. Am. Chem. Soc., 2013, 135, 1102–1109 CrossRef PubMed.
- K. G. Stamplecoskie, M. Grenier and J. C. Scaiano, J. Am. Chem. Soc., 2014, 136, 2956–2959 CrossRef CAS PubMed.
- J. Liu, M. Yu, C. Zhou, S. Yang, X. Ning and J. Zheng, J. Am. Chem. Soc., 2013, 135, 4978–4981 CrossRef CAS PubMed.
- K. Singh and S. Arora, Crit. Rev. Environ. Sci. Technol., 2011, 41, 807–878 CrossRef CAS.
- M. Rafatullah, O. Sulaiman, R. Hashim and A. Ahmad, J. Hazard. Mater., 2010, 177, 70–80 CrossRef CAS PubMed.
- V. K. Gupta, R. Kumar, A. Nayak, T. A. Saleh and M. A. Barakat, Adv. Colloid Interface Sci., 2013, 193–194, 24–34 CrossRef CAS PubMed.
- K. Ariga, A. Vinu, M. Miyahara, J. P. Hill and T. Mori, J. Am. Chem. Soc., 2007, 129, 11022–11023 CrossRef CAS PubMed.
- M. M. Naim and Y. M. El Abd, Sep. Purif. Rev., 2002, 31, 171–228 CrossRef CAS PubMed.
- S. S. Vaghela, A. D. Jethva, B. B. Mehta, S. P. Dave, S. Adimurthy and G. Ramachandraiah, Environ. Sci. Technol., 2005, 39, 2848–2855 CrossRef CAS.
- C. A. Martinez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105–145 CrossRef CAS PubMed.
- J. Gregory and J. Duan, Pure Appl. Chem., 2001, 73, 2017–2026 CrossRef CAS.
- K. Shi, M. Ren and I. Zhitomirsky, ACS Sustainable Chem. Eng., 2014, 2, 1289–1298 CrossRef CAS.
- K. Shi and I. Zhitomirsky, ACS Appl. Mater. Interfaces, 2013, 5, 13161–13170 CAS.
- M. R. Deakin and D. A. Buttry, Anal. Chem., 1989, 61, 1147A–1154A CrossRef CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- L. Proevska and I. Pojarlieff, Dyes Pigm., 1998, 36, 177–190 CrossRef CAS.
- C. Ying and H. Xi-Wen, Spectrochim. Acta, Part A, 1998, 54, 883–892 CrossRef.
- G. Sharma, M. Roy and S. Gupta, Synth. Met., 1997, 88, 57–63 CrossRef CAS.
- R. Jain, N. Sharma and K. Radhapyari, J. Appl. Electrochem., 2009, 39, 577–582 CrossRef CAS.
- A. Karunya, C. Rose and C. Valli Nachiyar, World J. Microbiol. Biotechnol., 2014, 30, 915–924 CrossRef CAS PubMed.
- P. Dachipally and S. B. Jonnalagadda, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2011, 46, 887–897 CrossRef CAS PubMed.
- K. A. Matis and G. P. Gallios, Int. J. Miner. Process., 1989, 25, 261–274 CrossRef CAS.
- H. Wang, A. C. Forse, J. M. Griffin, N. M. Trease, L. Trognko, P.-L. Taberna, P. Simon and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 18968–18980 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- F. W. Richey, B. Dyatkin, Y. Gogotsi and Y. A. Elabd, J. Am. Chem. Soc., 2013, 135, 12818–12826 CrossRef CAS PubMed.
- H. Wang, T. K. J. Koster, N. M. Trease, J. Segalini, P.-L. Taberna, P. Simon, Y. Gogotsi and C. P. Grey, J. Am. Chem. Soc., 2013, 133, 19270–19273 CrossRef PubMed.
- P. Simon and Y. Gogotsi, Acc. Chem. Res., 2012, 46, 1094–1103 CrossRef PubMed.
- M. D. Levi, G. Salitra, N. Levy, D. Aurbach and J. Maier, Nat. Mater., 2009, 8, 872–875 CrossRef CAS PubMed.
- J. N. Barisci, G. G. Wallace and R. H. Baughman, Electrochim. Acta, 2000, 46, 509–517 CrossRef CAS.
- S. Sigalov, M. D. Levi, G. Salitra, D. Aurbach and J. Maier, Electrochem. Commun., 2010, 12, 1718–1721 CrossRef CAS PubMed.
- N. Levy, M. D. Levi, D. Aurbach, R. Demadrille and A. Pron, J. Phys. Chem. C, 2010, 114, 16823–16831 CAS.
- M. D. Levi, N. Levy, S. Sigalov, G. Salitra, D. Aurbach and J. Maier, J. Am. Chem. Soc., 2010, 132, 13220–13222 CrossRef CAS PubMed.
- S. Sigalov, M. D. Levi, G. Salitra, D. Aurbach, A. Janes, E. Lust and I. C. Halalay, Carbon, 2012, 50, 3957–3960 CrossRef CAS PubMed.
- M. D. Levi, S. Sigalov, G. Salitra, R. Elazari and D. Aurbach, J. Phys. Chem. Lett., 2011, 2, 120–124 CrossRef CAS.
- W.-Y. Tsai, P.-L. Taberna and P. Simon, J. Am. Chem. Soc., 2014, 136, 8722–8728 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: BET results, cyclic voltammetry, chronopotentiometry, impedance spectroscopy data and cyclic behaviour, basic properties of dyes. See DOI: 10.1039/c4ra12635a |
| This journal is © The Royal Society of Chemistry 2015 |