
Pyrrolic nitrogen-doped carbon nanotubes: physicochemical properties, interactions with Pd and their role in the selective hydrogenation of nitrobenzophenone†
Lucy M. Ombaka,
Patrick G. Ndungu and
Vincent O. Nyamori*
School of Chemistry and Physics, University of KwaZulu-Natal, Westville Campus, Private Bag X54001, Durban, 4000, South Africa. E-mail: nyamori@ukzn.ac.za; Fax: +27 31 260 3091; Tel: +27 31 260 8256
First published on 24th November 2014
Abstract
Nitrogen-doped carbon nanotubes (N-CNTs) containing 63%, 73% and 80% pyrrolic-N were synthesized and used to evaluate the influence of pyrrolic nitrogen on the physicochemical properties and catalytic activity of Pd supported on N-CNTs (Pd/N-CNTs). Micrographs of Pd/N-CNTs showed that Pd was located along the defect sites of N-CNTs indicating strong Pd-support interactions. X-ray photoelectron spectroscopy revealed that the abundance of Pd0 decreased while that of Pd2+ increased as the quantity of pyrrolic nitrogen increased. The Pd2+ species were formed as Pd–N coordination complexes, which stabilized Pd2+ nanoparticles. Selective hydrogenation of nitrobenzophenone to aminobenzophenone or p-benzylaniline was used to evaluate the catalytic performance of catalysts. Pd/N-CNTs exhibited a higher selectivity towards aminobenzophenone than Pd on carbon nanotubes and Pd on activated carbon. The enhanced selectivity towards nitro-reduction alone, observed with Pd/N-CNTs was attributed to the promoting effect of pyrrolic-N. Hence, Pd/N-CNTs are promising catalysts for the selective reduction of nitro arenes.
1. Introduction
Graphene and carbon nanotubes (CNTs) are carbonaceous materials exhibiting unique properties, which make them attractive materials for use as metal catalyst supports.1,2 These properties include, high mechanical strength,3,4 good chemical stability,3,5 high surface area,6,7 notable electronic interactions with metal catalysts such as Pt and Ru8,9 and a mesoporous structure10 which facilitates favourable reactant-product mass transport.11 Due to these unique properties, CNTs and graphene are classified together with other smart carbonaceous supports such as graphene oxides and graphyne, which show novel properties as catalyst supports.12–14When used as metal catalyst supports, CNTs show good mechanical and chemical stability,3 metal dispersion,15 higher substrate conversion, and an improved regio- and stereoselectivity16 compared with traditional supports. This in turn has economic benefits since yields are generally increased and by-products or waste is reduced thus upholding green chemistry principles. Nonetheless, activated carbon (AC) is still a common carbonaceous material used as a catalyst support in the synthesis of industrial chemicals owing to its high surface area. However, AC suffers drawbacks such as faster deactivation rates,17 poor metal-support electronic interactions, lower thermal and mechanical stability, metal catalyst agglomeration and leaching.18 Thus, CNTs, which exhibit better support properties, are potentially the next generation of preferred carbonaceous supports.
Recent reports indicate that CNTs exhibit superior catalytic activity in industrial applications such as hydrogenation19 and dehydrogenation2 reactions. To improve the activity of CNTs, heteroatom-containing functional groups can be introduced onto the surface of CNTs. Heteroatoms such as nitrogen, oxygen, phosphorous, sulphur and boron can be used to modify the surface chemistry of CNTs.2 Incorporation of nitrogen functionalities into the graphene layers of CNTs to form nitrogen-doped CNTs (N-CNTs) improves their utility in catalytic reactions.20 For example, the presence of basic functional groups such as pyrrolic and pyridinic nitrogen in N-CNTs can enhance the chemical2 and electronic properties of N-CNTs.21 The improved chemical and electrical properties of N-CNTs result in activation of the metal catalyst22 and enhancement of the binding energy between N-CNTs and metal catalysts.23 Doping of CNTs with nitrogen also improves the physical properties of CNTs by introducing surface defects into the graphene structure of N-CNTs. Such surface defects promote N-CNTs surface wetting resulting in increased catalytic activity.24
Depending on the procedure used to synthesize N-CNTs, various nitrogen species such as pyridine-like, pyridine–N-oxide, pyrrole-like, carbonitrile, graphitic/quaternary nitrogen and lactams can be present in N-CNTs (Fig. 1). The different nitrogen species influence the catalytic activity of N-CNTs in diverse ways. For instance, pyridinic nitrogens were reported to enhance the catalytic activity of Pd supported on N-CNTs.25 However, the selective doping of N-CNTs with a single nitrogen species to facilitate structure–activity relationship studies remains under-researched.
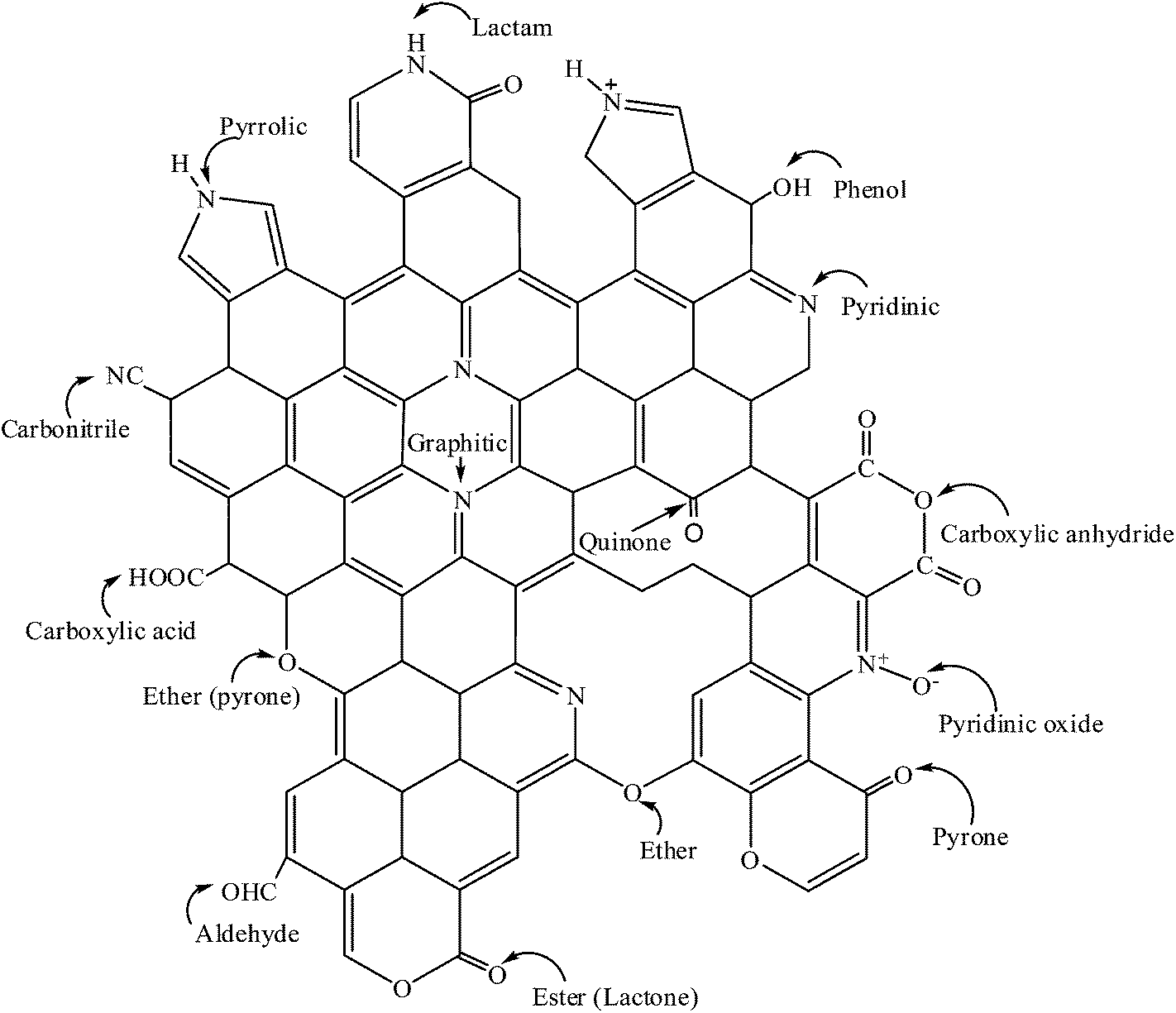 | ||
| Fig. 1 Graphical representation of possible nitrogen and oxygen functional groups present in N-CNTs. | ||
Oxygen functional groups can be added to the surface of N-CNTs via in situ or ex situ treatment of N-CNTs with oxygen containing organic or inorganic compounds.2 Oxygen functional groups such as carboxyl, carbonyl, lactone and quinone (Fig. 1) enhance the wettability of N-CNTs, further improving the catalytic performance of N-CNTs. The oxygen functional groups also act as anchoring sites for well dispersed metal nanoparticles supported on N-CNTs. Thus, compared with CNTs, oxygen-functionalized N-CNTs exhibit enhanced metal nanoparticle properties such as better stability, smaller particle size, better dispersion and activation, which results in higher catalytic activity and selectivity.2 As a result, oxygen-functionalised N-CNTs are potentially smart materials for application as catalysts or as metal catalyst supports used in catalytic reactions.
The catalytic activity of CNTs in hydrogenation reactions has been considerably studied.19 However, limited studies report the catalytic activity of N-CNTs in general, and more specifically pyrrolic containing N-CNTs in the selective hydrogenation of nitro-arenes. Selective hydrogenation of nitro-arenes to amino-arenes is of immense industrial importance as amino-arenes are used in pharmaceuticals and in the production of fine chemicals. Aminobenzophenone is an example of an amino-arene which is industrially important as it is used as an intermediate in the synthesis of Schiff bases,26 and in nonlinear optical materials generating blue lasers.27,28 p-Benzylaniline is another amino-arene that has the potential to be used as an intermediate for the synthesis of other Schiff bases. In this study, the selective hydrogenation of nitrobenzophenone (NBP) to aminobenzophenone was chosen as a model reaction. This is because NBP contains two reducible functional groups (NO2 and CO), which allows for the evaluation of catalysts' selectivity towards reduction of the nitro group alone.
Ferrocene is commonly used as a catalyst in the CVD synthesis of N-CNTs.29 To modulate the level of nitrogen-doping and the nitrogen species incorporated into N-CNTs, nitrogen-containing ferrocenyl derivatives can be used as catalysts instead of ferrocene.30 In this study, the effect of using 3-ferrocenyl-2-(4-cyanophenyl)acrylonitrile as a catalyst for the introduction of nitrogen species into N-CNTs was evaluated. We report the selective synthesis of pyrrolic N-CNTs and their physicochemical properties. The influence of pyrrolic nitrogen on the chemical properties of Pd nanoparticles supported on the pyrrolic N-CNTs is also reported. Additionally, we investigated the role of pyrrole-like nitrogen in the selective hydrogenation of nitrobenzophenone to aminobenzophenone over Pd supported on N-CNTs. For comparison purposes, the catalytic activity of Pd supported on CNTs and also on AC was evaluated under similar reactions.
2. Experimental
2.1. Materials and instrumentation
The reagents 4-nitrobenzophenone (99%), 4-aminobenzophenone (97%) and diethyl ether (99.8%) were purchased from Sigma Aldrich, Germany. Palladium acetylacetonate {Pd(acac)2} (synthesis grade) was purchased from Merck, Germany. Pd on activated carbon (5 wt%) was purchased from Sigma-Aldrich, Germany. Toluene (99.4%) was purchased from LiChroSolv, Germany. All reagents and solvents were of analytical grade and used as received from the suppliers. Pd was loaded onto N-CNTs and CNTs in a cylindrical stainless steel reaction chamber (140 mm × 10 mm) sealed at one end with a Swagelok® fitting nut. To monitor the partial pressure used during synthesis of the catalyst, a Thyracont VD84/1 Pirani vacuum gauge was used. Hydrogenation of 4-nitrobenzophenone was conducted in a Parr® Instrument Co. 4848 reactor. The catalyst was separated from the reactants by first filtering the mixture through Whatman ashless, no. 42 filter paper purchased from Sigma-Aldrich Germany, followed by filtration through a PVDF 0.45 μm (GVS) membrane syringe filters purchased from Lasec, SA.2.2. Synthesis of N-CNTs and CNTs
N-CNTs were synthesized by use of a chemical vapour deposition (CVD) method similar to that reported by Oosthuizen et al.31 and Koch et al.32 Briefly, the compound 3-ferrocenyl-2-(4-cyanophenyl)-acrylonitrile that we previously synthesized and characterized (ESI S1†), was used as a catalyst, and it was synthesized following the protocol outlined by Imrie et al.33 and Ombaka et al.34 Acetonitrile was used as a carbon and nitrogen source, while ethylbenzoate was used as a source of oxygen. The solution used to synthesize N-CNTs was prepared by dissolving 0.25 g of the catalyst in 9.75 g of acetonitrile to make a total of 10 g of solution (i.e. 2.5 wt% of catalyst). To introduce 1 wt% and 2 wt% of oxygen into the synthesis precursors, 0.5 and 1.0 g of ethylbenzoate was added to 0.25 g of the catalyst respectively. Each solution was then prepared to a total mass of 10 g with acetonitrile. The solution of precursors was injected into a quartz tube (placed inside a tube furnace) at a flow rate of 0.8 mL min−1 with a syringe pump. The injected precursors were carried through the quartz tube by a carrier gas made-up of 10% hydrogen in argon (v/v) which was pumped through the system at a rate of 100 mL min−1 and a pressure of 80 kPa. The furnace was set to a reaction temperature of 850 °C for 30 minutes.CNTs were also synthesized by a CVD method as previously detailed by Oosthuizen et al.31 For synthesis of CNTs, ferrocene was used as the catalyst while toluene was used as the carbon source. The solution used to synthesize CNTs was made by dissolving 0.25 g of ferrocene in 9.75 g of toluene to make 10 g of solution. The CNTs were synthesized at a temperature of 850 °C for 30 minutes by use of similar synthesis conditions as those outlined for N-CNTs.
The N-CNTs and CNTs were collected from the hot zone of the quartz tube upon completion of the reaction. All products were purified by first calcining the samples at 300 °C for 3 hours, followed by ultrasonication of the samples in 6 M nitric acid for 40 minutes at room temperature. The sonicated mixture was then refluxed at 100 °C for 24 hours at a constant stirring rate of 300 rpm. After refluxing, the acid was neutralized with 3 M NaOH and the mixture sonicated for 40 minutes. Afterwards, the N-CNTs or CNTs were separated from the mixture via filtration and washed with deionized water until a neutral pH was obtained.
2.3. Synthesis of catalysts
Synthesis of the catalysts was achieved by loading Pd onto acid treated N-CNTs and CNTs to yield Pd/N-CNTs and Pd/CNTs respectively. Loading of 5 wt% Pd onto each support was achieved via a metal organic-CVD (MOCVD) method as outlined by Suttisawat et al.35 In detail, 0.072 g of Pd(acac)2 was mixed with 0.475 g of acid treated N-CNTs or CNTs and ground thoroughly by using a pestle and mortar. The resulting mixture was placed inside a stainless steel MOCVD reactor and the reactor components sealed. The sealed MOCVD reactor was connected to a vacuum pump maintained at a partial pressure of 2.2 × 10−2 mbar for 45 minutes. After evacuating the MOCVD reactor for 45 minutes, it was placed in the middle of a muffle furnace operated at 120 °C for 30 minutes. This was followed by increasing the temperature to 300 °C at a rate of 2 °C min−1 and thereafter maintaining it at 300 °C for 45 minutes. The system was then allowed to cool to ambient temperature while still under vacuum. Upon completion of the reaction, the formed catalysts were removed from the reactor by scraping with a spatula, and then characterized as outlined in Section 2.5.2.4. Catalytic tests
Hydrogenation of nitrobenzophenone to aminobenzophenone was conducted in a closed vessel reactor by using a mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :66 (Pd:nitrobenzophenone). For all catalytic tests, 180.0 mg of the catalyst was put into a Parr reactor stainless steel vessel and mixed with 1250 mg of 4-nitrobenzophenone and 100 mL of dry toluene. The reactor vessel and its contents was sealed and then stirred continuously under hydrogen gas at the desired reaction conditions. A control experiment without any catalyst was performed under similar conditions. To monitor the reaction progress, aliquots of 1 mL of the reactant mixture were collected from the reaction vessel at intervals of 2 hours, quenched with 4 mL of toluene, filtered as described in Section 2.1 and then analysed off-line by GC-FID or GC-MS. Upon completion of the reaction, the solution in the reaction vessel was cooled to ambient temperature, filtered as described in Section 2.1 and the filtrate kept in a sealed container for further analysis. The used catalyst was obtained as a filtration residue, washed with 100 mL of diethylether, dried at 100 °C overnight in an oven, and then kept in a sealed container for X-ray diffraction analysis. The conversion of 4-nitrobenzophenone (NBP) and selectivity to 4-aminobenzophenone (ABP) was calculated by using eqn (A) and (B), respectively.
:66 (Pd:nitrobenzophenone). For all catalytic tests, 180.0 mg of the catalyst was put into a Parr reactor stainless steel vessel and mixed with 1250 mg of 4-nitrobenzophenone and 100 mL of dry toluene. The reactor vessel and its contents was sealed and then stirred continuously under hydrogen gas at the desired reaction conditions. A control experiment without any catalyst was performed under similar conditions. To monitor the reaction progress, aliquots of 1 mL of the reactant mixture were collected from the reaction vessel at intervals of 2 hours, quenched with 4 mL of toluene, filtered as described in Section 2.1 and then analysed off-line by GC-FID or GC-MS. Upon completion of the reaction, the solution in the reaction vessel was cooled to ambient temperature, filtered as described in Section 2.1 and the filtrate kept in a sealed container for further analysis. The used catalyst was obtained as a filtration residue, washed with 100 mL of diethylether, dried at 100 °C overnight in an oven, and then kept in a sealed container for X-ray diffraction analysis. The conversion of 4-nitrobenzophenone (NBP) and selectivity to 4-aminobenzophenone (ABP) was calculated by using eqn (A) and (B), respectively.
 | (A) |
 | (B) |
2.5. Characterization
Images of N-CNTs, CNTs and all catalysts were taken by using a transmission electron microscope (TEM) (JEOL JEM 1010) and a scanning electron microscope (SEM) (JEOL JSM 6100). A high resolution-TEM (JEOL 2100) was used to take higher magnification images. The thermalstability of N-CNTs and CNTs was determined by using a Q Series™ Thermal Analyzer DSC/TGA (Q600). X-ray photoelectron spectroscopy (XPS) analysis was performed with a KRATOS AXIS Ultra DLD equipped with Al Kα (1486 eV) X-rays, an X-ray Power of 20 W and a beam diameter of 100 μm. Fourier transform infrared spectra (FTIR) of supports and catalysts embedded into KBr pellets were recorded on a Perkin Elmer spectrum RX1 FTIR spectrometer. Temperature programmed reduction (TPR) analysis was conducted in 5% H2 in argon by using a Micromeritics Autochem II Chemisorption Analyzer (2920). The graphitic nature of N-CNTs and CNTs was determined by use of a Raman spectrometer (DeltaNu Advantage 532™). XRD spectra were obtained from a Bruker D8 Advance X-ray diffractometer equipped with a graphite monochromatized high-intensity Cu Kα radiation (λ = 0.15406 nm). The Pd-loading content was determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) (Perkin Elmer Optima 5300 DV). Details for ICP-OES analysis are provided in the ESI S1.† The surface area and nitrogen adsorption–desorption isotherms of the catalysts were determined by using a Micromeritics Tristar II surface area and porosity analyser. The hydrogenation products were characterized by using GC-FID (Shimadzu 2010 gas chromatograph) and GC-MS (Shimadzu GCMS-QP2010 SE gas chromatograph-mass spectrometer). The conditions under which the GC-FID and GC-MS analysis were performed are provided in ESI S1.†3. Results and discussion
3.1. Characterization of N-CNTs and CNTs
 | ||
| Fig. 2 TEM images of (A) CNTs, (B) N-CNTs-0 and (C) N-CNTs-2. | ||
All N-CNTs exhibited a reduced outer diameters (OD) compared with those of CNTs (Fig. 3). The reduced OD can be explained by the fact that, nitrogen species dominate tube edges impeding further formation of graphitic layers and resulting in closure of the tube.36 The OD of N-CNTs-1 and N-CNTs-2 were observed to be smaller than that of N-CNTs-0. Since N-CNTs-1 and N-CNTs-2 were synthesized using 1 and 2 wt% oxygen, the further reduction of their OD can be ascribed to the etching effect of oxygen resulting in thinner tube walls and smaller OD.
 | ||
| Fig. 3 Outer diameter and inner diameter of N-CNTs and CNTs; for each sample at least 80 tubes were measured. | ||
All N-CNTs exhibited larger inner diameters (ID) compared with those of CNTs (Fig. 3). This can be attributed to the formation of larger iron catalyst nanoparticles (NPs) in N-CNTs compared to those formed in CNTs during synthesis. It is possible that, the heteroatom-containing ferrocenyl derivative used to synthesize N-CNTs predominantly forms larger iron catalyst NPs compared to those formed by ferrocene which was used to synthesize CNTs.38 Another possible explanation is that the presence of nitrogen and oxygen heteroatoms during synthesis facilities formation of larger iron catalyst NPs.39 The presence of bigger catalyst NPs in N-CNTs can be related to the increase in residual iron content (CNTs < N-CNTs-0 < N-CNTs-1 < N-CNTs-2) evidenced from the (thermalgravimetric analysis) TGA thermograms of as-synthesized samples (Fig. 4A). The bigger catalyst particles could have favoured formation of larger inner diameters.
 | ||
| Fig. 4 Thermograms of (A) as-synthesized and (B) purified CNTs and N-CNTs; derivative thermograms of (C) as-synthesized and (D) purified CNTs and N-CNTs. The symbols hash (#) and asterisk (*) denote the as-synthesized and purified samples respectively. | ||
All purified N-CNTs were less thermally stable than CNTs implying that the purification procedure did not severely remove the nitrogen present in N-CNTs. The thermal stability of purified N-CNTs decreased in the order N-CNTs-2 > N-CNTs-1 > N-CNTs-0. This decrease in thermal stability was attributed to an increase in pyrrole-like nitrogen. Purification of N-CNTs and CNTs reduced the iron content in all samples to similar quantities (Fig. 4B). A significant reduction of the peak associated with amorphous carbons at ca. 400 °C, marked by a dotted circle was achieved after sample purification (cf. Fig. 4C and D).
3.2. Characterization of catalysts
The N 1s spectra of all N-CNTs showed a broad peak between 399–405 eV resulting from an overlap of different peaks (Fig. 5A). Deconvolution of the broad N 1s peak gave three peaks at 398.5–398.4, 400.3–400.6 and 404.1–404.7 eV (ESI S3†). The peak at 398.4–398.5 eV was attributed to pyridinic and carbonitrile nitrogen species and it was coded N1.42,43 The peak at 400.3–400.6 eV was accredited to pyrrole nitrogen species and it was coded N2.43–45 While the peak at 404.1–404.7 eV was attributed to nitrogen molecules adsorbed or encapsulated within the N-CNTs, it was coded N3.46
 | ||
| Fig. 5 (A) N 1s XPS spectra of N-CNTs synthesized by using 0–2 wt% oxygen in acetonitrile, and (B) variations in the nitrogen species present in N-CNTs synthesized by using 0–2 wt% oxygen. | ||
Increasing the amount of oxygen from 0–2 wt% during the synthesis of N-CNTs, resulted in an increase in the amount of N2 and a decrease in the amount of N1 and N3 (Fig. 5B). This could imply that the presence of oxygen favoured formation of N2 at the expense of N1 and N3. The decrease in N3 can be due to formation of NOx molecules via the reaction between oxygen and nitrogen molecules. The NOx molecules exit the reaction chamber as exhaust gases. On the other hand, the decrease in N1 was accredited to an additional reaction induced by the presence of oxygen that favoured formation of N2. The possibility of an additional reaction initiated by oxygen was deduced from evaluating the C 1s and O 1s spectra of N-CNTs and CNTs.
Deconvolution of the C 1s spectra of N-CNTs and CNTs gave two common carbon species (C1 and C2) in all N-CNTs and CNTs. A third carbon species (C3) was only present in N-CNTs-1 and N-CNTs-2, while the fourth carbon species (C4) was only present in CNTs and N-CNTs-0 (Table 1 and ESI S3†). The lowest energy peak obtained was at 284.4–284.7 eV (C1); this was assigned to graphitic carbons.42,47 A second peak was obtained at 285.5–285.9 eV (C2); this peak was assigned to C–N and C–O species.48,49 The third peak was obtained at 287.3–287.7 eV (C3); this was attributed to carbonyls in quinone, lactones and carboxyl groups.49,50 The fourth peak was obtained at 288.2–288.8 eV (C4); this was ascribed to carbonyls in carboxyl groups.49,50 Resonance shake-up satellite peaks caused by π–π transitions in the aromatic rings were observed at 291.1–291.6 eV.50
| Sample | C at.% | O at.% | |||
|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | O Total | |
| Pd/N-CNTs-0 | 52.7 | 33.8 | — | 8.9 | 10.4 |
| Pd/N-CNTs-1 | 45.9 | 28.6 | 21.7 | — | 8.3 |
| Pd/N-CNTs-2 | 42.4 | 27.3 | 23.5 | — | 13.5 |
| Pd/CNTs | 57.4 | 25.1 | — | 10.0 | 6.6 |
All N-CNTs exhibited lower quantities of C1 (graphitic carbon) compared with CNTs implying that, nitrogen-doping reduced the graphitic nature of CNTs. Additionally, the amount of graphitic carbon in N-CNTs further reduced as the percentage of pyrrolic groups increased (cf. Table 1 and Fig. 5B). Since the quantity of graphitic carbon in N-CNTs did not show a positive correlation with the total nitrogen percentage, the decrease in graphitic carbon was ascribed to formation of more pyrrole groups. An increase in pyrrolic groups enhanced the disorder in the N-CNTs, accounting for the reduced thermal stability of N-CNTs-1 and N-CNTs-2 observed from the thermograms and DTG of these samples. Similarly, the decrease in graphitic carbon resulted in decreased crystallinity of the N-CNTs as observed from Raman analysis51 (ESI S1†).
Based on the C 1s and O 1s spectra of N-CNTs, we propose that during the synthesis of N-CNTs-1 and N-CNTs-2 lactones are formed52 as explained in the ESI S4.† The formed lactones react with ammonia obtained from the reaction of hydrogen and acetonitrile53 to form lactams. The lactams are then converted to pyrrole groups by the removal of a –CO group (Scheme 1).44 As more oxygen is added into the synthesis precursor, more lactones are formed resulting in an increased formation of pyrrole groups at the expense of pyridinic and carbonitrile groups. Hence, lactones provided an additional route to that already present in N-CNTs-0, by which pyrrole groups are formed, thus, increasing selectivity towards pyrrole formation.
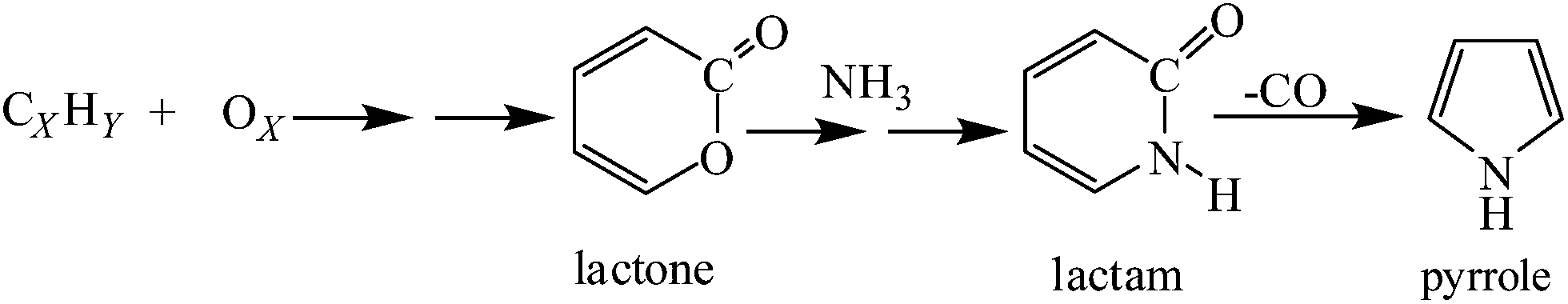 | ||
| Scheme 1 Plausible synthetic route for pyrrole formation. | ||
Further identification of the functional groups present in Pd/N-CNTs and Pd/CNTs was accomplished by the use of FTIR. From the FTIR spectra of all samples, a clear distinction was observed between the functional groups present in N-CNTs and CNTs (Fig. 6). At ≈2373 cm−1 a peak caused by an overlap between carbonitriles and C–O stretching vibrations was observed.54,55 The intensity of this peak was strong in Pd/N-CNTs but very weak in Pd/CNTs. Hence, in Pd/N-CNTs the peak at ≈2373 cm−1 was majorly from carbonitrile groups, further supporting the presence of carbonitriles in N-CNTs as observed from XPS analysis. Since the peak at ≈2373 cm−1 was weak in Pd/CNTs, it is possible that in Pd/CNTs this peak originated only from C–O stretching vibrations.
 | ||
| Fig. 6 FTIR spectra of Pd/CNTs and Pd/N-CNTs. | ||
Another important peak was that at 1466 cm−1 assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in pyridinic groups.55,56 The intensity of this peak was quite weak in all Pd/N-CNTs but completely absent in Pd/CNTs. The transmittance peak of CC in the graphene layers of CNTs was observed at 1653 cm−1,57,58 while the peak assigned to C–N groups was observed at ≈1570 cm−1.55,59 In Pd/CNTs, the peaks attributed to C–N and carbonitrile groups were absent but they were clearly visible in Pd/N-CNTs. The absence of these peaks indicated that no nitrogen-doping was present in Pd/CNTs as also observed from XPS analysis (ESI S3†). It was interesting to note that for N-CNTs-0 the CC and C–N peaks exhibited almost the same intensity while in N-CNTs-1 and N-CNTs-2 the intensity of the CC peak was significantly lower than that of C–N. This was related to the presence of more pyrrole groups in N-CNTs-1 and N-CNTs-2 compared with N-CNTs-0.
N in pyridinic groups.55,56 The intensity of this peak was quite weak in all Pd/N-CNTs but completely absent in Pd/CNTs. The transmittance peak of CC in the graphene layers of CNTs was observed at 1653 cm−1,57,58 while the peak assigned to C–N groups was observed at ≈1570 cm−1.55,59 In Pd/CNTs, the peaks attributed to C–N and carbonitrile groups were absent but they were clearly visible in Pd/N-CNTs. The absence of these peaks indicated that no nitrogen-doping was present in Pd/CNTs as also observed from XPS analysis (ESI S3†). It was interesting to note that for N-CNTs-0 the CC and C–N peaks exhibited almost the same intensity while in N-CNTs-1 and N-CNTs-2 the intensity of the CC peak was significantly lower than that of C–N. This was related to the presence of more pyrrole groups in N-CNTs-1 and N-CNTs-2 compared with N-CNTs-0.
The peak at 3420 cm−1, attributed to hydrogen bonded –OH groups,60,61 was present in all samples. Peaks at 1393 and 1258 cm−1 allotted to stretching of the C–O and CC in the tubes61 were stronger in Pd/CNTs than in Pd/N-CNTs. While the peaks at 1089 and 790 cm−1, assigned to C–O vibrations and stretching modes,57,58 were only present in Pd/CNTs. The carbonyl peak appearing at 1706 cm−1 was very weak in Pd/CNTs but slightly stronger in Pd/N-CNTs.57,58 This could imply that the hydrogen bonded –OH groups in Pd/CNTs were mainly from phenol groups and adsorbed water, while the –OH groups in Pd/N-CNTs are mainly due to carboxylic groups and adsorbed water. The peak at 2937 cm−1, assigned to CH2 groups, was present in all samples.55,57
| Catalyst | At.% N | Pyrrole composition (%) | Surface area of catalyst (m2 g−1) | Pore size distribution (nm) |
|---|---|---|---|---|
| Pd/CNTs | 0 | 0 | 52 | 4.1 |
| Pd/N-CNTs-0 | 4.4 | 63 | 55 | 4.5 |
| Pd/N-CNTs-1 | 3.5 | 73 | 89 | 4.1 |
| Pd/N-CNTs-2 | 2.5 | 80 | 38 | 4.1 |
All supports were classified as type IV mesoporous materials, exhibiting a hysteresis loop between 0.5–1.0 P/P0 (Fig. 7, ESI S4†).62 Pd/CNTs and Pd/N-CNTs-0 showed a hysteresis loop between 0.7–1.0 P/P0 which appeared very similar to a H1 type hysteresis loop.63 Such a hysteresis loop is intrinsic of independent regular cylindrical and spherical shaped pores.64 In addition, Pd/N-CNTs-0 showed a more open hysteresis loop than Pd/CNTs (ESI S4†), indicating that the pore size of CNTs increased upon doping with nitrogen (Table 2). The increase in pore size observed with Pd/N-CNTs-0 was associated with the increase of ID observed from TEM analysis (Fig. 3).
 | ||
| Fig. 7 Representative N2 adsorption–desorption isotherms of Pd/CNTs and Pd/N-CNTs. | ||
Pd/N-CNTs-1 and Pd/N-CNTs-2 showed an almost horizontal hysteresis loop between 0.5–0.9 P/P0 with a cavitation at ca. 0.5 P/P0 that is associated with a H3 type hysteresis loop.63 The H3 type hysteresis loop could be due to disordered pores forming the network of a porous matrix,65 or large pores between entangled nanotubes. The disordered pores could be due to extensive structural disorders dominated with pyrrole groups, which are five membered rings that distort the graphene structure of CNTs. Alternatively, the disordered pores could result from oxygen etching some of the formed regular shaped pores, making them irregularly shaped.
 | ||
| Fig. 8 Pd 3d XPS spectra of Pd/CNTs and Pd/N-CNTs synthesized by using 0–2% oxygen in acetonitrile. | ||
| Pd 3d5/2 | Pd 3d3/2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample | Pd0 | Pd2+ | Pd0 | Pd2+ | ||||
| BE (eV) | At.% | BE (eV) | At.% | BE (eV) | At.% | BE (eV) | At.% | |
| Pd/N-CNTs-0 | 335.6 | 36.5 | 337.7 | 20.5 | 340.8 | 24.8 | 342.8 | 18.1 |
| Pd/N-CNTs-1 | 335.4 | 20.0 | 337.3 | 34.1 | 340.8 | 23.1 | 342.8 | 22.7 |
| Pd/NCNTs-2 | 335.2 | 8.16 | 337.0 | 45.2 | 340.3 | 11.3 | 342.4 | 29.3 |
| Pd/CNTs | 335.6 | 44.11 | 337.5 | 11.5 | 340.9 | 44.11 | 342.9 | 10.62 |
As shown in Table 3 the binding energies at ca. 335.6 and 340.8 eV were assigned to Pd0 species47 while, those at ca. 337.7 and 342.8 eV were assigned to Pd2+ species.66–69 The Pd2+ species in Pd/CNTs could be PdO formed via an oxidation reaction between Pd0 and the surface oxygen groups in CNTs. However, the Pd2+ species in N-CNTs were not entirely from PdO as no linear correlation was observed between the oxygen percentage and the percentage of Pd2+ (cf. Tables 1 and 3). It was noted that as the amount of pyrrolic nitrogen increased from 0 (in CNTs) to 80% (in N-CNTs-2), the quantity of Pd0 reduced while that of Pd2+ increased (Table 3 and Fig. 8). This indicated that as the percentage of pyrrolic nitrogen increased, Pd0 species derived from Pd(acac)2 were progressively being bonded to the pyrrolic nitrogen to form Pd–N complexes. This interaction could occur during synthesis of the catalyst following the reaction depicted in Scheme 2. Such a reaction could be due to the interaction of Pd0 with neighbouring pyrrolic cations.66,70 The covalently bonded Pd2+ could be stable, preventing further reduction of Pd2+ to Pd0 during synthesis.
 | ||
| Scheme 2 Possible interactions between pyrrolic nitrogens and Pd nanoparticles. | ||
To probe further, the existence of an interaction between Pd NPs and N-CNTs, the FTIR spectrum of N-CNTs-2 was compared with that of Pd/N-CNTs-2 (Fig. 9A). In N-CNTs-2 the peak assigned to C–N groups appeared at ca. 1567 cm−1, while in Pd/N-CNTs-2 this peak appears at around 1552 cm−1.59 The shift of the C–N peak to lower wavenumbers in the presence of Pd NPs is indicative of interactions between pyrrolic nitrogen and Pd NPs.70,71
 | ||
| Fig. 9 (A) FTIR spectra of N-CNTs-2 and Pd/N-CNTs-2, and (B) TPR profiles of Pd/CNTs and Pd/N-CNTs. | ||
The interaction between Pd NPs and the surface of different supports was also studied by using TPR and the profiles obtained are presented in Fig. 9B. From the TPR profiles, a broad peak was observed between 60–150 °C; this peak was associated with reduction of Pd2+ species, possibly in PdO.72 The Pd2+ reduction peak was observed to gradually broaden towards higher temperatures as the quantity of pyrrolic groups increased in N-CNTs-0 and N-CNTs-2. This shift towards higher temperatures is indicative of formation of stronger metal-support interactions as the number of pyrrolic nitrogen groups increased. However, the Pd2+ reduction peak of Pd/N-CNTs-1 occurred at a lower temperature than expected.
TPR profiles showed that the intensity of the Pd2+ reduction peak did not increase in a manner similar to that observed from the Pd 3d XPS spectra (Fig. 8). This implied that the increased quantity of Pd2+ observed from the Pd 3d XPS spectra was not entirely due to the formation of more PdO. This observation further supported our hypothesis that the increase in Pd2+ species in Pd/N-CNTs was due to interactions between Pd NPs and pyrrolic nitrogen groups. A weak desorption peak was observed at ca. 50 °C in the TPR profile of Pd/CNTs alone. This peak was ascribed to decomposition of Pd-β-hydride73 and its absence in all the other profiles implied that no Pd-β-hydride was formed when Pd NPs were supported on N-CNTs.
The TEM images of Pd/CNTs and Pd/N-CNTs are presented in Fig. 10. Pd NPs supported on N-CNTs-0, N-CNTs-1 and N-CNTs-2 exhibited a more even particle dispersion compared with Pd NPs supported on CNTs (cf. Fig. 10A–D). Pd NPs were randomly distributed on CNTs, while on N-CNTs-1 and N-CNTs-2 Pd NPs were typically located along the defect sites. These defect sites consist of pentagons and heptagons induced by nitrogen-doping.36 This indicated that electronic charges are localized on the pentagons and heptagons, which enhance electronic interactions between Pd NPs and defect sites, leading to well dispersed Pd NPs.74,75 Interestingly, hexagonal Pd NPs were only observed in Pd/N-CNTs-2 as shown in Fig. 10E which is a higher magnification of Fig. 10D. This could imply that the high composition of pyrrolic nitrogen (80%) provided a sufficient quantity of molecules that acted as surface capping agents, also facilitated formation of hexagonal Pd NPs.
 | ||
| Fig. 10 TEM images of (A) Pd/CNTs, (B) Pd/N-CNTs-0, (C) Pd/N-CNTs-1, (D) Pd/N-CNTs-2 and (E) a higher magnification image of Pd/N-CNTs-2. Inset histograms in images a, b, c and d are the corresponding Pd NPs size distributions. | ||
Representative images of supported Pd NPs analysed by HR-TEM are shown in Fig. 11. From the HR-TEM images, Pd NPs supported on N-CNTs-0 (Fig. 11A) appeared spherical while Pd NPs supported on N-CNTs-2 (Fig. 11B) appeared hexagonal. The hexagonal Pd NPs similar to those observed in the lower magnification images of Pd/N-CNTs-2 (Fig. 10E).
 | ||
| Fig. 11 TEM images of (A) Pd/CNTs-0 and (B) Pd/N-CNTs-2. | ||
High magnification images of Pd/N-CNTs-0 showed the presence of tiny Pd NPs forming nanoclusters and larger Pd NPs (Fig. 11A). Such tiny particles were mainly present in Pd/N-CNTs-0 containing the highest level of nitrogen-doping. Consequently, formation of nanoclusters was ascribed to greater nitrogen-doping levels and the presence of more pyridinic/carbonitrile groups. Generally, higher nitrogen-doping levels favoured the formation of tiny Pd NPs, while higher levels of pyrrolic nitrogen favoured formation of hexagonal Pd NPs.
The XRD diffractograms of various catalysts are presented in Fig. 12 where Pd/AC was used as an external standard. The peak at 26.4° was indexed as the C(002) reflection of the hexagonal graphite in CNTs and N-CNTs. The peaks at 40° and 45° present in Pd/AC, Pd/CNTs and all Pd/N-CNTs were indexed as the Pd (111) and Pd (200) reflections of Pd0.76 Pd/CNTs showed an extra peak at 67° that was indexed as the Pd (220) reflection. All catalysts had similar Pd loading levels as obtained from ICP-OES analysis (Table 4). Therefore, the absence of the Pd (220) reflection in Pd/N-CNTs was related to differences in Pd NP sizes. A further evaluation of the Pd NP sizes was performed by using the Pd (111) diffraction peak and TEM image analysis.
 | ||
| Fig. 12 XRD diffractograms of Pd/CNTs, Pd/N-CNTs and Pd/AC. | ||
| Catalyst | Pd loading wt% (ICP-OES) | Ave. Pd NP size (nm) TEM | Ave. Pd NP diameter (nm) XRD | 2θ of Pd(111) peak (°) | FWHM of Pd(111) peak (°) | Inter layer spacing (Å) |
|---|---|---|---|---|---|---|
| a NP: nanoparticle.b As determined by the manufacturer, Ave: average, FWHM: full width at half maximum. | ||||||
| Pd/CNTs | 4.9 | 8 | 4.5 | 39.429 | 1.867 | 2.28347 |
| Pd/N-CNTs-0 | 4.5 | 5 | 3.8 | 39.938 | 2.254 | 2.25554 |
| Pd/N-CNTs-1 | 5.0 | 7 | 4.0 | 39.924 | 2.11 | 2.25631 |
| Pd/N-CNTs-2 | 4.9 | 5 | 4.1 | 39.892 | 2.085 | 2.25807 |
| Pd/AC | 5.0b | 12 | 10.7 | 40.278 | 0.793 | 2.23728 |
The Pd NP sizes were estimated from the Pd (111) diffraction peak, by applying the Debye-Scherrer equation given by:

In addition, the particle size of supported Pd NPs was determined by measuring the size of at least 200 Pd NPs from the TEM images (Fig. 10). Both the TEM and XRD analyses of Pd NP sizes showed a comparable trend in the particle size variation (Table 4). Based on the XRD analysis, Pd NPs supported on CNTs exhibited larger particle sizes than Pd NPs supported on N-CNTs. Pd NPs supported on N-CNTs-0 showed the smallest particle size. The Pd NP sizes decreased as the total percentage nitrogen in N-CNTs increased. This indicated that increasing nitrogen-doping levels in N-CNTs introduced more functional groups which adequately anchored the Pd NPs. The well-anchored Pd NPs were less prone to NP sintering and agglomeration during the cooling stages of catalyst synthesis resulting in smaller particle sizes.2
3.3. Catalytic activity tests
The catalytic activity of Pd/N-CNTs, Pd/CNTs, N-CNTs-2 and Pd/AC was evaluated with the aim of determining the influence of pyrrolic nitrogen on the selective nitro-reduction of NBP. Since NBP has three reducible groups vis. NO2, CHO and CC, selectivity towards ABP was more vital than the conversion of NBP because this selectivity is an indication of the catalysts' efficiency in the formation of substituted anilines. Fig. 13 and Table 5 presents the catalytic activity of these catalysts. All Pd/N-CNTs showed a greater conversion of NBP than Pd/CNTs after 24 hours of the reaction. This indicated that doping of CNTs with nitrogen enhanced the activity of Pd NPs (Fig. 13A).
 | ||
| Fig. 13 (A) Conversion of NBP over Pd/N-CNTs, Pd/CNTs, Pd/AC and (B) conversion of NBP over Pd/N-CNTs-2 and N-CNTs-2. | ||
| Catalyst | Conversion of NBP (%) | Selectivity to ABP (%) | Selectivity to p-benzylaniline (%) | Other productsa |
|---|---|---|---|---|
| a Other hydrogenation products detected from the GC-MS. | ||||
| Pd/CNTs | 85 | 70 | — | Morpholine-4-carbal, 2-phenylethanol |
| Pd/N-CNTs-0 | 93 | 63 | 6.3 | Morpholine-4-carbal |
| Pd/N-CNTs-1 | 100 | 83 | — | Morpholine-4-carbal |
| Pd/N-CNTs-2 | 98 | 89 | — | Morpholine-4-carbal |
| Pd/AC | 100 | 24 | 24 | Benzophenone |
Pd/N-CNTs-1 and Pd/N-CNTs-2 with a smaller at.% N content were more active than Pd/N-CNTs-0 which had the highest at.% N. Since Pd/N-CNTs-1 and Pd/N-CNTs-2 had a higher pyrrolic N content than Pd/N-CNTs-0, it is plausible that pyrrolic nitrogen enhances activity. The pyrrolic nitrogen groups could have promoted Pd-support interactions by forming Pd–N coordination complexes.77 Such complexes can alter the electronic and structural properties of Pd NPs as explained in Section 3.2.3 resulting in improved activities.76 The improved activity of Pd/N-CNTs could also be due to the stronger metal-support interactions as observed from TPR analysis. Such strong metal-support interactions can enhance Pd NP dispersion, reduce Pd NP agglomeration and sintering during reactions, resulting in a higher activity.25
Pd/N-CNTs-2 with the highest percentage of pyrrolic nitrogen and the lowest surface area showed an activity higher than that of Pd/N-CNT-0 and Pd/CNTs, but slightly lower than that of Pd/N-CNTs-1. To further probe the cause of the enhanced activity over Pd/N-CNTs-2, the activity of purified N-CNTs-2 was evaluated. N-CNTs-2 exhibited a higher activity than Pd/N-CNTs-2 (Fig. 13A). A possible explanation for the lower activity observed with Pd/N-CNTs-2 is that some of the pyrrolic groups were bound to Pd NPs, thus reducing the quantity of pyrrolic groups available for NBP conversion. This implied that the pyrrolic groups in N-CNTs-2 largely promoted the catalytic activity of Pd/N-CNTs-2. Thus, the enhanced activity of Pd/N-CNTs-2 despite having a low surface area, could be due to the promotin effect of pyrrolic groups. The availability of more pyrrolic groups also resulted in more disorders in Pd/N-CNTs-2 compared with other catalysts as evidenced form the XPS and TGA analysis. Such a high content of disordered sites are related to nitrogen-induced defects which can lower the work function of materials resulting in increased catalyst activity.78 Pd/AC showed the highest activity, and this was attributed to its high surface area of 890 m2 g−1 (ESI S5†). No correlation was observed between Pd NP size and catalytic activity.
Hydrogenation of NBP over Pd/CNTs, Pd/N-CNTs-1 and Pd/N-CNTs-2 yielded only ABP, while hydrogenation of NBP over Pd/N-CNTs-0 and Pd/AC yielded ABP and p-benzylaniline (Scheme 3). Although, Pd/AC showed the highest convertion of NBP, its selectivity to either ABP or p-benzylaniline was poor compared to that of Pd/CNTs and Pd/N-CNTs. Up to 10 hours of reaction, all Pd/N-CNTs showed greater selectivity towards ABP than Pd/CNTs (Fig. 14A and Table 5). Thereafter selectivity over Pd/N-CNTs-2 increased, and that of Pd/N-CNTs-1 and Pd/CNTs were almost constant while that of Pd/N-CNTs-0 sharply dropped. Pd/AC showed the lowest selectivity to ABP throughout the whole reaction period. Given that Pd/-N-CNTs-1 and Pd/N-CNTs-2 showed a greater selectivity than Pd/CNTs, we deduced that nitrogen-doping of CNTs enhanced the selective reduction of the nitro group to an amine group.79 As the amount of pyrrolic nitrogen increased, selectivity towards ABP also increased for Pd/N-CNTs-1 and Pd/N-CNTs-2, while no correlation was observed between the selectivities and the total N%. This implied that selectivity was influenced more by the presence of pyrrolic nitrogen than the total N%. Other studies have shown that the species of nitrogen and not just the nitrogen content in N-CNTs play a significant role in influencing the catalytic properties of N-CNTs as metal catalyst supports.80
 | ||
| Scheme 3 Hydrogenation of NBP over various catalysts. | ||
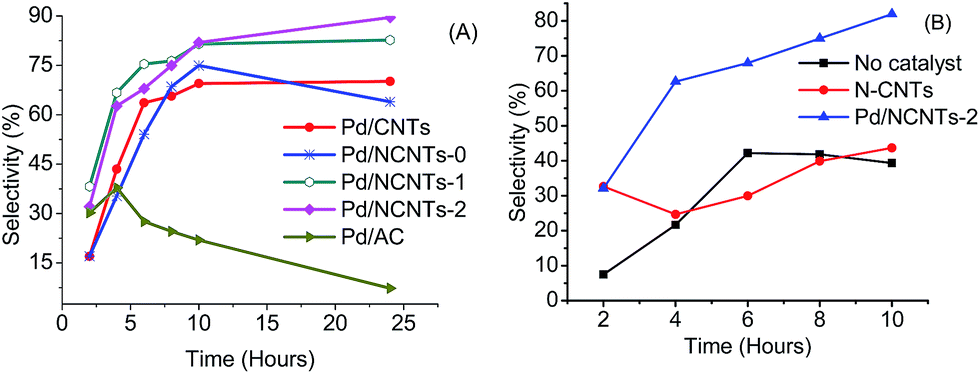 | ||
| Fig. 14 (A) Selectivity towards ABP over Pd/N-CNTs, Pd/CNTs, Pd/AC and (B) selectivity towards ABP over Pd/N-CNTs-2 and N-CNTs-2. | ||
N-CNTs-2 showed a much lower selectivity towards ABP compared with Pd/N-CNTs-2 (Fig. 14B), implying that Pd NPs were responsible for the selectivity observed with Pd/N-CNTs-2. Therefore, the enhanced selectivity over Pd/N-CNTs was majorly attributed to the presence of Pd–N complexes. Such complexes minimise Pd NP sintering and agglomeration during the reaction.76 This increases the stability and dispersion of Pd NPs while minimizing the formation of Pd black which is catalytically inactive. The increased stability and dispersion of Pd NPs could increase the hydrogen chemisorption capacity of Pd NPs hence increasing the selectivity. The presence of more disordered pores in N-CNTs could also facilitate a faster mass transfer of ABP from the catalyst surface back into the solution.2 This could prevent a further reduction of ABP to p-benzylaniline resulting in an increased selectivity. Corma et al. compared the selective nitro-reduction of nitrobenzaldehyde to aminobenzaldehyde over supported Au and Pd.81 Their findings shows a lower selectivity over Pd compared with Au. Hence, the enhanced selectivity towards ABP observed with Pd/NCNTs can be due to the promoting effect of N-CNTs supports.
Selectivity over Pd/N-CNTs-0 dropped after 10 hours implying that Pd/N-CNTs-0 possess a different catalytic pathway compared with Pd/CNTs, Pd/N-CNTs-1 and Pd/N-CNTs-2. The different catalytic pathway in Pd/N-CNTs-0 was accredited to the existence of tiny Pd NPs forming nanoclusters. A similar drop in selectivity was observed over Pd/AC which also had tiny Pd NPs in the form of clusters (ESI S6†). Such Pd nanoclusters may possess a different reaction mechanism from the larger Pd NPs.82 This different reaction mechanism could have favoured the further reduction of ABP to p-benzylaniline accounting for the drop in selectivity (ESI S6† and Table 5).
Since all catalysts had a similar quantity of residual iron catalyst (Fig. 4B) the possibility of the residual iron catalyst influencing the observed catalytic trend was ruled out. From the TEM analysis no iron particles were observed on the surface of the supports. This implied that the residual iron was encapsulated inside the CNTs and N-CNTs and hence not available for surface catalysis.
Pd/N-CNTs-2, which showed the highest selectivity after 24 hours, was used to evaluate the effect of pressure on the catalytic activity and selectivity. It was observed that as the pressure increases the catalytic activity and selectivity over Pd/N-CNTs-2 reduced (ESI S7†). Thus, the optimum pressure for the hydrogenation of NBP was 1 bar.
3.4. Stability of Pd NPs after hydrogenation reactions
The stability of Pd NPs on N-CNTs-0, N-CNTs-1 and N-CNTs-2 after use was analysed by XRD. From the XRD diffractograms, the Pd NP size of used catalysts was determined in a similar manner to that outlined in Section 3.2.3. Used Pd/CNTs was not analysed because it strongly adhered to the filter paper resulting in a low catalyst recovery, which was insufficient for XRD analysis. Table 6 shows the average diameters of the Pd NPs before and after reaction. The size of the Pd NPs in N-CNTs-0 and N-CNTs-1 increased after the reaction while in N-CNTs-2 the size of the Pd NPs did not change. This implied that Pd sintering and agglomeration may have occurred only in Pd/N-CNTs-0 and Pd/N-CNTs-1. Thus, the Pd NPs in N-CNTs-2 were considered very stable; owing to the formation of more Pd–N complexes in Pd/N-CNTs-2 than in Pd/N-CNTs-0 and Pd/N-CNTs-1. In a similar manner, Chen et al. showed that pyridinic nitrogen can complex with Ru3+ as a ligand to form stable Ru3+ particles, which were resilient to metal sintering and agglomeration.20The diffractograms of the used catalysts showed an increase in the intensity of the Pd (111) reflection compared with those of the fresh catalyst (ESI S8†). This increase in the Pd (111) reflection indicated that more Pd0 were formed via the hydrogen reduction of Pd2+ during the reaction.
4. Conclusions
Pyrrolic N-CNTs containing 2.5–4.4 at.% of nitrogen were successfully synthesized by means of CVD method. Selectivity towards formation of pyrrolic nitrogen was promoted by the incorporation of oxygen derived from ethylbenzoate during synthesis of the N-CNTs. Increasing the amounts of pyrrolic groups in N-CNTs significantly altered the physicochemical properties of N-CNTs such as thermal stability, textural properties and surface wetness. Also, an increase in the pyrrolic percentage improved the interactions between Pd and N-CNTs resulting in better dispersion and more stable Pd NPs. The improved dispersion and stability of Pd NPs was associated with a strong interaction between the pyrrolic nitrogen and Pd NPs. The catalytic activity and selectivity of Pd/N-CNTs in the hydrogenation of NBP was dependent on the percentage of pyrrolic nitrogen and not on the total nitrogen content. Pd/N-CNTs showed an enhanced selectivity compared with Pd/CNTs and Pd/AC. Thus, Pd supported on pyrrolic N-CNTs is a promising catalyst for the selective reduction of NBP and other industrially important nitro arenes such as nitrobenzene derivatives.Acknowledgements
The authors wish to thank the University of KwaZulu-Natal and the National Research Foundation (NRF) for financial support. We are grateful to Professor Bice Martincigh and Dr R. S. Mwakubambanya for their insightful suggestions during the manuscript development and their assistance with proof-reading.References
- Y. Tang, Z. Yang, X. Dai, D. Ma and Z. Fu, J. Phys. Chem. C, 2013, 117, 5258–5268 CAS.
- L. M. Ombaka, P. Ndungu and V. O. Nyamori, Catal. Today, 2013, 217, 65–75 CrossRef CAS PubMed.
- H. Dai, Nanotube growth and characterization, in Carbon Nanotubes Synthesis, Structure, Properties and Applications, ed. M. S. Dresselhaus and G. Dresselhaus, Springler-Verlag, Heidelberg, 2001, pp. 29–51 Search PubMed.
- A. Politano, A. R. Marino, D. Campi, D. Farías, R. Miranda and G. Chiarello, Carbon, 2012, 50, 4903–4910 CrossRef CAS PubMed.
- J. S. Qi, J. Y. Huang, J. Feng, D. N. Shi and J. Li, ACS Nano, 2011, 5, 3475–3482 CrossRef CAS PubMed.
- A. Loiseau, X. Blasé, J.-C. Charlier, P. Gadelle, C. Journet, C. Laurent and A. Peigney, Synthesis methods and growth mechanism, in Understanding Carbon Nanotubes, ed. A. Loiseau, P. Launois, P. Petit, S. Roche and J.-P. Salvetat, Springer-Heidelberg, Springer, 2006, pp. 49–122 Search PubMed.
- C. Wang, L. Zhang, Z. Guo, J. Xu, H. Wang, K. Zhai and X. Zhuo, Microchim. Acta, 2010, 169, 1–6 CrossRef CAS.
- J. Wang, G. Yin, Y. Shao, Z. Wang and Y. Gao, J. Phys. Chem., 2008, 112, 5784–5789 CAS.
- A. Politano and G. Chiarello, Nanoscale, 2013, 5, 8215–8220 RSC.
- J.-S. Lee, S.-I. Kim, J.-C. Yoon and J.-H. Jang, ACS Nano, 2013, 7, 6047–6055 CrossRef CAS PubMed.
- J. J. Niu, J. N. Wang, Y. Jiang, L. F. Su and J. Ma, Microporous Mesoporous Mater., 2007, 100, 1–5 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- D. Malko, C. Neiss, F. Viñes and A. Görling, Phys. Rev. Lett., 2012, 108, 086804–086807 CrossRef.
- Y. Wang, N. Shah, F. E. Huggins and G. P. Huffman, Energy Fuels, 2006, 20, 2612–2615 CrossRef CAS.
- J.-H. Olivier, F. Camerel, R. Ziessel, P. Retailleau, J. Amadou and C. Pham-Huu, New J. Chem., 2008, 32, 920–924 RSC.
- M. P. Lázaro, E. García-Bordeje, D. Sebastián, M. J. Lázaro and R. Moliner, Catal. Today, 2008, 138, 203–209 CrossRef PubMed.
- S. Domínguez-Domínguez, Á. Berenguer-Murcia, B. K. Pradhan, Á. Linares-Solano and D. Cazorla-Amorós, J. Phys. Chem. C, 2008, 112, 3827–3834 Search PubMed.
- R. S. Oosthuizen and V. O. Nyamori, Platinum Met. Rev., 2011, 55, 154–169 CrossRef CAS.
- J. J. L. Chen, Z. H. Zhu, S. B. Wang, Q. Ma, V. Rudolph and G. Q. Lu, Chem. Eng. J., 2010, 156, 404–410 CrossRef CAS PubMed.
- E. M. M. Ibrahim, O. K. Vyacheslav, A. Leonhardt, S. Hampel, S. Oswald, M. H. Rümmeli and B. Büchner, Diamond Relat. Mater., 2010, 19, 1199–1206 CrossRef CAS PubMed.
- F. R. García-García, J. Àlvarez-Rodríguez, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Carbon, 2010, 48, 267–276 CrossRef PubMed.
- W. An and C. H. Turner, J. Phys. Chem. C, 2009, 113, 7069–7078 CAS.
- C. Yonghai, Y. Hao, T. Jun, P. Feng, W. Hongjuan, L. Jing, Z. Wenxu and W. Ning-Bew, Carbon, 2013, 57, 433–442 CrossRef PubMed.
- K. Chizari, I. Janowska, M. Houllé, I. Florea, O. Ersen, T. Romero, P. Bernhardt, M. J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2010, 380, 72–80 CrossRef CAS PubMed.
- A. S. Saghiyan, L. A. Stepanyan, L. L. Manasyan, A. V. Geolchanyan, S. M. Djamgaryan, H. R. Ajvazyan, H. A. Panosyan, V. I. Maleev and T. F. Saveleva, Tetrahedron Lett., 2010, 21, 2638–2645 CrossRef CAS PubMed.
- A. K. Bhowmik, S. Tan, A. C. Ahyi, J. A. Dharmadhikari, A. K. Dharmadhikari and D. Mathur, Opt. Commun., 2007, 280, 472–476 CrossRef CAS PubMed.
- Z. Li, B. Wu, G. Su and G. Huang, Appl. Phys. Lett., 1997, 70, 562–564 CrossRef CAS PubMed.
- E. N. Nxumalo and N. J. Coville, Materials, 2010, 3, 2141–2171 CrossRef CAS PubMed.
- E. N. Nxumalo, V. O. Nyamori and N. J. Coville, J. Organomet. Chem., 2008, 693, 2942–2948 CrossRef CAS PubMed.
- R. S. Oosthuizen and V. O. Nyamori, Appl. Organomet. Chem., 2012, 26, 536–545 CrossRef CAS.
- R. J. Koch, M. Weser, W. Zhao, F. Viñes, K. Gotterbarm, S. M. Kozlov, O. Höfert, M. Ostler, C. Papp, J. Gebhardt, H. P. Steinrück, A. Görling and T. Seyller, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 075401–075406 CrossRef.
- C. Imrie, P. Kleyi, V. O. Nyamori, T. I. A. Gerber, D. C. Levendis and J. Look, J. Organomet. Chem., 2007, 692, 3443–3453 CrossRef CAS PubMed.
- L. M. Ombaka, P. G. Ndungu, B. Omondi and V. O. Nyamori, J. Coord. Chem., 2014, 67, 1905–1922 CrossRef CAS.
- Y. Suttisawat, P. Rangsunvigit, B. Kitiyanan, M. Williams, P. Ndungu, M. V. Lototskyy, A. Nechaev, V. Linkov and S. Kulprathipanja, Int. J. Hydrogen Energy, 2009, 34, 6669–6675 CrossRef CAS PubMed.
- B. G. Sumpter, V. Meunier, J. M. Romo-Herrera, E. Cruz-Silva, D. A. Cullen, H. Terrones, D. J. Smith and M. Terrones, ACS Nano, 2007, 1, 369–375 CrossRef CAS PubMed.
- L. Lizhao, L. Feng and Z. Jijun, Nano Res., 2014, 7, 626–657 CrossRef PubMed.
- K. Chizari and U. Sundararaj, Mater. Lett., 2014, 116, 289–292 CrossRef CAS PubMed.
- S. Liu, Y. Zhang, Y. Lin, Z. Zhao and Q. Li, Carbon, 2014, 69, 247–254 CrossRef CAS PubMed.
- K. Chizari, A. Vena, L. Laurentius and U. Sundararaj, Carbon, 2014, 68, 369–379 CrossRef CAS PubMed.
- A. A. Koós, M. Dowling, K. Jurkschat, A. Crossley and N. Grobert, Carbon, 2009, 47, 30–37 CrossRef PubMed.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- E. T. Kang, K. G. Neoh, S. H. Khor, K. L. Tan and B. T. G. Tan, Polymer, 1990, 31, 202–207 CrossRef CAS.
- S. Kundu, W. Xia, W. Busser, M. Becker, D. A. Schmidt, M. Havenith and M. Muhler, Phys. Chem. Chem. Phys., 2010, 12, 4351–4359 RSC.
- S. Wang, F. Wang and X. Ge, Synth. Met., 1986, 16, 99–104 CrossRef CAS.
- Y. Cao, H. Yu, J. Tan, F. Peng, H. Wang, J. Li, W. Zheng and N.-B. Wong, Carbon, 2013, 57, 433–442 CrossRef CAS PubMed.
- C. J. Powell, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 1–3 CrossRef CAS PubMed.
- S. C. Ray, A. Saha, N. R. Jana and R. Sarkar, J. Phys. Chem. C, 2009, 113, 18546–18551 CAS.
- D. Briggs and G. Beamson, Anal. Chem., 1992, 64, 1729–1736 CrossRef CAS.
- S. Kundu, Y. Wang, W. Xia and M. Muhler, J. Phys. Chem. C, 2008, 112, 16869–16878 CAS.
- S. Z. Mortazavi, P. Parvin, A. Reyhani, R. Malekfar and M. Soghra, RSC Adv., 2013, 3, 1397–1409 RSC.
- G. Beamson, D. T. Clark, N. W. Hayes and D. S.-L. Law, Surf. Sci. Spectra, 1994, 3, 357–365 CrossRef CAS.
- H. Li, Y. Wu, H. Luo, M. Wang and Y. Xu, J. Catal., 2003, 214, 15–25 CrossRef CAS.
- A. Misra, P. K. Tyagi, M. K. Singh and D. S. Misra, Diamond Relat. Mater., 2006, 15, 385–388 CrossRef CAS PubMed.
- G. Lazar and I. Lazar, J. Non-Cryst. Solids, 2003, 331, 70–78 CrossRef CAS PubMed.
- T. Barzetti, E. Selli, D. Moscotti and L. Forni, J. Chem. Soc., Faraday Trans., 1996, 92, 1401–1407 RSC.
- L. Vanyoreka, R. Meszarosa and S. Barany, Colloids Surf., A, 2014, 448, 140–146 CrossRef PubMed.
- P. E. Fanning and M. A. Vannice, Carbon, 1993, 31, 721–730 CrossRef CAS.
- T. J. Bandosz, Carbon materials fo catalysis, John Wiley & Sons, Inc., United States of America, 2009, pp. 63–66 Search PubMed.
- U.-J. Kim, C. A. Furtado, X. Liu, G. Chen and P. C. Eklund, J. Am. Chem. Soc., 2005, 127, 15437–15445 CrossRef CAS PubMed.
- M. Aydin, Vib. Spectrosc., 2013, 65, 84–93 CrossRef CAS PubMed.
- Z. Lei, D. Bai and X. S. Zhao, Microporous Mesoporous Mater., 2012, 147, 86–93 CrossRef PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- C. J. Rasmussen, A. Vishnyakov, M. Thommes, B. M. Smarsly, F. Kleitz and A. V. Neimark, Langmuir, 2010, 26, 10147–10157 CrossRef CAS PubMed.
- H.-J. Woo and P. A. Monson, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 041207–041224 CrossRef.
- J. Y. Kim, K. Park, S. Y. Bae, G. C. Kim, S. Lee and H. C. Choi, J. Mater. Chem., 2011, 21, 5999–6005 RSC.
- X.-F. Guo, D.-Y. Jang, H.-G. Jang and G.-J. Kim, Catal. Today, 2012, 186, 109–114 CrossRef CAS PubMed.
- S. Santra, P. Ranjan, P. Bera, P. Ghosh and S. K. Mandal, RSC Adv., 2012, 2, 7523–7533 RSC.
- T. H. Fleisch and G. J. Mains, J. Phys. Chem., 1986, 90, 5317–5320 CrossRef CAS.
- I. Rivas, J. Badano, C. Lederhos, D. Liprandi, E. Cagnola and C. V. M. Quiroga, Quim. Nova, 2011, 34, 87–90 CrossRef CAS PubMed.
- M. Baghayeri, H. Veisi, H. Veisi, B. Maleki, H. Karimi-Malehd and H. Beitollahi, RSC Adv., 2014, 4, 49595–49604 RSC.
- O. S. G. P. Soares, J. J. M. Órfão and M. F. R. Pereira, Ind. Eng. Chem. Res., 2010, 49, 7183–7192 CrossRef CAS.
- M. Gurratha, T. Kuretzky, H. P. Boehm, L. B. Okhlopkova, A. S. Lisitsyn and V. A. Likholobov, Carbon, 2000, 38, 1241–1255 CrossRef.
- A. Staykov, Y. Ooishi and T. Ishihara, J. Phys. Chem. C, 2014, 118, 8907–8916 CAS.
- L. Bardotti, F. Tournus, R. Delagrange, J. M. Benoit, O. Pierre-Louis and V. Dupuis, Appl. Surf. Sci., 2014, 301, 564–567 CrossRef CAS PubMed.
- P. Chen, L. M. Chew, A. Kostka, M. Muhlera and W. Xia, Catal. Sci. Technol., 2013, 3, 1964–1971 CAS.
- D. He, C. Zeng, C. Xu, N. Cheng, H. Li, S. Mu and M. Pan, Langmuir, 2011, 27, 5582–5588 CrossRef CAS PubMed.
- Q. B. Wen, L. Qiao, W. T. Zheng, Y. Zeng, C. Q. Qu, S. S. Yu and Q. Jiang, Phys. E, 2008, 40, 890–893 CrossRef CAS PubMed.
- P. Chen, F. Yang, A. Kostka and W. Xia, ACS Catal., 2014, 4, 1478–1486 CrossRef CAS.
- W. Ouyang, D. Zeng, X. Yu, F. Xie, W. Zhang, J. Chen, J. Yan, F. Xie, L. Wange, H. Meng and D. Yuan, Int. J. Hydrogen Energy, 2014, 39, 15996–16005 CrossRef CAS PubMed.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- C. Torres, C. Campos, J. L. G. Fierro, M. Oportus and P. Reyes, Catal. Lett., 2013, 143, 763–771 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12523a |
| This journal is © The Royal Society of Chemistry 2015 |