
A facile synthesis approach to micro–macroporous carbon from cotton and its application in the lithium–sulfur battery†
Hongqiang Wanga,
Zhixin Chenc,
Hua Kun Liua and
Zaiping Guo*abc
aInstitute for Superconducting & Electronic Materials, University of Wollongong, NSW 2522, Australia. E-mail: zguo@uow.edu.au
bHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, College of Chemistry and Chemical Engineering, Hubei University, Wuhan 430062, P. R. China
cSchool of Mechanical, Materials & Mechatronics Engineering, University of Wollongong, NSW 2500, Australia
First published on 24th November 2014
Abstract
A new type of low-cost activated micro–macroporous carbon suitable for mass production that is derived from cotton was successfully prepared by using potassium hydrate in a chemical activation method. The activated carbon exhibits a hierarchically porous microstructure and high specific surface area (1286 m2 g−1). The micro–macroporous structure allows a large amount of sulfur (68%) to be infiltrated into the micropores of the host. When evaluated as a cathode for lithium–sulfur batteries, the hierarchically porous carbon–sulfur composite electrode exhibits excellent cycling stability and good performance. The resulting composite electrode possesses a reversible capacity of 760 mA h g−1 after 200 cycles at the 0.2 C current rate.
1. Introduction
The lithium–sulfur battery is expected to help meet the demand for power sources for electric vehicles due to its abundant raw materials, nontoxicity, and high theoretical storage capacity (1675 mA h g−1).1–4 Practical application of the Li–S battery, however, is greatly hampered by two major challenges. One is the low electrical conductivity of sulfur and the intermediate polysulfide products.5–7 The other one is the high solubility of the intermediate lithium polysulfides in organic electrolytes, which can shuttle between the anode and cathode, resulting in the precipitation of solid Li2S2 and Li2S on the cathode.8–10 These issues lead to the low utilization of sulfur, low coulombic efficiency, and severe degradation of cycle life.4,11In attempts to overcome the problems mentioned above, various strategies have been developed, such as optimization of the organic electrolyte3,12–16 and the preparation of conductive polymer/sulfur composites17–20 and carbon/sulfur composites. It should be noted that employing porous carbon to constrain the sulfur or lithium polysulfide within a framework, such as microporous carbon,21–27 mesoporous carbon,28–32 hollow carbon spheres,33,34 porous carbon fibers,35 carbon nanotiles,36 or carbon nanotubes37,38 can effectively minimize the shuttling of polysulfides and increase the utilization and cycling stability of sulfur cathodes. Carbon substrates with different pore types can offer different advantages. Macroporous carbon can store more sulfur in its large pores, however, the macropores only weakly confine polysulfides, and the macroporous carbon exhibits inferior electrochemical properties. Compared with their macroporous counterpart, both microporous carbon and mesoporous carbon exhibit much improved sulfur utilization and sulfur retention, leading to good electrochemical performance. The disadvantage is that the sulfur content in the composite is low. In addition, the majority of carbon materials used in confining sulfur so far are difficult to apply in practical applications due to their complicated preparation and processing, and their high cost.
Cotton is well known as one of the most important and common commodities in the world. Selecting cotton as the precursor to prepare porous carbon can not only reduce its preparation costs and simplify its preparation process but can also achieve mass production. Herein, we report a novel and facile approach to synthesize cotton-based porous carbon, and use it as an improved confinement matrix to encapsulate active sulfur for high performance lithium–sulfur batteries.
2. Experimental
2.1 Preparation of porous carbon–sulfur composite
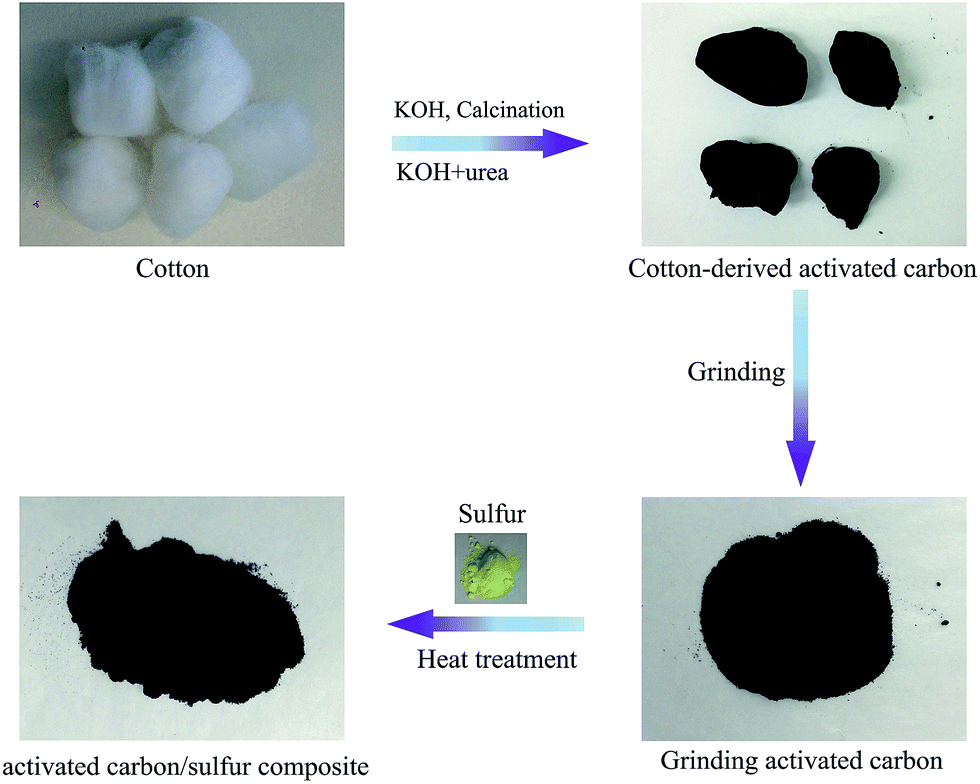
A schematic illustration of the preparation of porous carbon–sulfur composites from cotton is shown in Fig. 1. Firstly, 12 g cotton was impregnated with solvent mixtures of urea–KOH–H2O (U–K) (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:80 w/w) and KOH–H2O (K) (20:80) for 6 h, respectively. Then, each sample was dried at 80 °C for 24 h. The as-prepared precursor was then heated at 700 °C for 2 h under N2 atmosphere with the heating rate of 5 °C min−1. The resultant porous carbon (PC) samples were denoted as PC–U–K and PC–K, respectively. Actually, we designed a series of experiments to investigate the cotton treatment under different conditions and found that the adjustment of mass ratio of KOH to urea could control the nanostructure of porous carbon (see ESI, Fig. S1†). We found that the as-prepared samples with urea only or only a low concentration of KOH had lower surface area. Lower surface area and pore volume are not suitable for high S loading. Therefore, we chose two samples with different structures to discuss (one treated with 10% urea and 10% KOH, mainly showing micropores; the other one treated with 20% KOH, which is rich in micropores and macropores). The as-prepared porous carbon and sulfur were mixed together in a weight ratio of 2:8 and heated to 160 °C in a sealed stainless steel autoclave for 24 h to facilitate sulfur diffusion into the carbon host. Then, the composite was heated at 200 °C and kept for 10 minutes under flowing argon gas (50 cm3 s−1) to vaporize the sulfur deposited on the outside surface of the composite (with the samples at this stage denoted as PC–K–S and PC–U–K–S, respectively).
:10:80 w/w) and KOH–H2O (K) (20:80) for 6 h, respectively. Then, each sample was dried at 80 °C for 24 h. The as-prepared precursor was then heated at 700 °C for 2 h under N2 atmosphere with the heating rate of 5 °C min−1. The resultant porous carbon (PC) samples were denoted as PC–U–K and PC–K, respectively. Actually, we designed a series of experiments to investigate the cotton treatment under different conditions and found that the adjustment of mass ratio of KOH to urea could control the nanostructure of porous carbon (see ESI, Fig. S1†). We found that the as-prepared samples with urea only or only a low concentration of KOH had lower surface area. Lower surface area and pore volume are not suitable for high S loading. Therefore, we chose two samples with different structures to discuss (one treated with 10% urea and 10% KOH, mainly showing micropores; the other one treated with 20% KOH, which is rich in micropores and macropores). The as-prepared porous carbon and sulfur were mixed together in a weight ratio of 2:8 and heated to 160 °C in a sealed stainless steel autoclave for 24 h to facilitate sulfur diffusion into the carbon host. Then, the composite was heated at 200 °C and kept for 10 minutes under flowing argon gas (50 cm3 s−1) to vaporize the sulfur deposited on the outside surface of the composite (with the samples at this stage denoted as PC–K–S and PC–U–K–S, respectively).
 | ||
| Fig. 1 Schematic illustration of the preparation of porous carbon–sulfur composite. | ||
2.2 Characterization
The crystal phases and morphologies of the obtained materials were analyzed by powder X-ray diffraction (XRD) using an X-ray diffractometer (MMA GBC, Australia), by Raman spectroscopy on an instrument (JOBIN YVON HR800) equipped with a 632.8 nm diode laser, by JEOL JSM-7500FA field-emission scanning electron microscopy (FESEM), and by JEOL 2011 transmission electron microscopy (TEM). The content of sulfur in composites was measured by thermogravimetric analysis/differential scanning calorimetry (TGA/DSC) type instrument (METTLER TOLEDO, Switzerland) at a heating rate of 10 °C min−1 from room temperature to 500 °C under argon atmosphere. The porous structure of the samples was confirmed by N2 adsorption–desorption isotherms at 77 K using Micromeritics ASAP 2020 analyzer. Before the measurement, the samples were degassed at 110 °C for 24 h. The specific surface area was calculated by the Brunauer–Emmett–Teller (BET) method from the adsorption data in the relative pressure range (P/P0) of 0.05 to 0.2. The pore size distribution was analyzed by using the Horvath–Kawazoe (H–K) model.2.3 Electrochemical measurements
The PC–K–S and PC–U–K–S composites (80% by weight) were mixed with super-P (10%) and PVDF (10%) in NMP to form the slurry, respectively. After that, the electrodes were prepared by coating the slurry onto aluminum foil and then dried at 50 °C for 24 h. Coin-type (CR2032) cells were assembled in an argon-filled glove box. The electrolyte used was 1 M lithium bis(trifluoromethanesulfonyl)imide in a solvent mixture of 1,3-dioxolane (DOL):dimethoxyethane (DME) (1:1, v/v), containing LiNO3 (1 wt%). The coin cells were galvanostatically charged–discharged from 1.8 to 2.6 V using a cell test instrument (CT2001A, LAND, China).
3. Results and discussion
SEM images of the raw cotton and the porous carbon samples made from various conditions are shown in Fig. 2. The fibrous structure of the cotton was still maintained in the PC–U–K, while the fibrous structure was destroyed in the PC–K sample, which shows a three-dimensional porous structure. There are decomposition and drastic reaction between the carbon and KOH when the cotton is impregnated in KOH solution followed with carbonization process. These reactions results in the formation of macropores with pore size from 100 to 300 nm. In this case, KOH first reacts with carbon according to the reaction (1)| 6KOH + 2C = 2K + 3H2 + 2K2CO3 (when annealing temperature is above 400 °C) | (1) |
 | ||
| Fig. 2 SEM images of (a) raw cotton, (b) PC–U–K, (c) PC–K. | ||
After the reaction (1), the decomposition of K2CO3 and reactions of K/K2CO3/CO2 with carbon could generate a large amount of nanoscale pores.39 When the cotton was impregnated with KOH and urea simultaneously, the extent of the reaction between KOH and carbon was weakened, therefore, no obvious macropores were generated and the entire structure of cotton could be maintained. Indeed, the role of urea is to generate micropores as well, by decomposing to NH3 during the carbonization process. The yield of PC–U–K and PC–K are 15% and 9% based on the initial cotton weight, respectively.
In order to investigate the microstructure of the samples in detail, TEM and HRTEM were conducted at various magnifications. Fig. 3 shows TEM images of PC–U–K and PC–K. Compared to the PC–U–K, the PC–K sample shows hierarchically porous structure, and macropores are well distributed in the PC–K sample, which are connected with each other to form into a three-dimensional conductive network. It is not very easy to observe micropores from TEM images, however, and therefore, nitrogen adsorption isotherms were employed to further investigate the microporous structure. It can be imagined that the hierarchically porous structure with micro–macropores framework is highly desirable for high-performance electrode materials. The micro–macropores can not only facilitate loading of sulfur into the pores, but also provide a large reaction surface area, which helps electrolyte immersion and fast ion transport in the electrode/electrolyte interface, thus enhancing the electrochemical performance of electrodes.
 | ||
| Fig. 3 TEM images of (a)–(c) PC–U–K and (d)–(f) PC–K. | ||
Fig. 4 shows the nitrogen adsorption isotherms of the PC–U–K and PC–K. Both the PC–U–K and the PC–K present type-I isotherms, indicating the microporous feature of the samples. Moreover, the pore-size distributions of the carbon materials is shown in the Fig. 4 inset. The PC–U–K shows a dominant peak centered at 0.8 nm, and a peak centered at 0.9 nm can be observed for the PC–K. The BET surface area and total pore volume for PC–U–K and PC–K are 1286 and 1078 m2 g−1, and 1.15 and 0.87 cm3 g−1, respectively. The difference in the specific surface area and pore volume may be introduced by the macropores in the PC–K which generated by KOH treatment. These additional macropores of the PC–K sample compared to that of the PC–U–K sample will play a significant role in facilitating sulfur loading into micropores, fast transport of ion and electrolyte, consequently improving the electrochemical performance of sulfur/carbon cathode.
 | ||
| Fig. 4 N2 sorption isotherms of the PC–U–K and PC–K. Inset shows the pore-size distributions for the materials. | ||
The XRD patterns for the PC–K, S, and PC–K–S are shown in Fig. 5(a). The peak at 24.4° can be assigned to the (002) crystallographic planes of graphite in the PC–K sample. The low intensity and the high peak broadening are typical feature of amorphous carbon. In contrast, the pure elemental sulfur has well-defined diffraction peaks, corresponding to an orthorhombic structure. Although there is a substantial amount of sulfur (68%, see ESI, Fig. S2†) in the PC–K–S composite, the peaks of elemental sulfur were not observed, suggesting good dispersion of the S within the porous carbon host.22,40 Raman spectra of PC–K, S, and PC–K–S composite were also carried out to further observe their structure in Fig. 5(b). Element sulfur displays typical Raman peaks corresponding to the S–S bond,41 however, any sulfur peaks can not be observed in the PC–K–S, which is also indicates that sulfur is successfully embedded into the pores of the PC–K. The sulfur content in the PC–U–K–S and PC–K–S composites was determined by TGA conducted under argon atmosphere (see ESI, Fig. S2†). The amount of sulfur in the PC–U–K–S and PC–K–S composites is 50 wt% and 68 wt%, respectively.
 | ||
| Fig. 5 XRD patterns (a) and Raman spectra (b) of the PC–K, S, and PC–K–S composite. | ||
Fig. 6(a) and (b) displays the discharge–charge voltage profiles of the PC–U–K–S and PC–K–S electrodes for selected cycles. Typical plateaus are observed, which are characteristic of sulfur-based electrodes. The discharge plateau of the PC–U–K–S composite electrode obviously shrinks as the increase of cycle number, however, the PC–K–S electrode has a good overlap of discharge plateaus during the cycling tests, suggesting the excellent stability and reversibility of the PC–K–S electrode. After the 100th cycle, the retained discharge capacity for the PC–U–K–S composite electrode is 710 mA h g−1, indicating a poor cycling performance. In contrast, the capacity of the PC–K–S composite electrode capacity only drops to 850 mA h g−1 after the 100th cycle, with the retention rate of 83%. The cycling performances of the PC–U–K–S and PC–K–S are shown in Fig. 6(c). The initial discharge capacities for the PC–U–K–S and PC–K–S cathodes are 1020 and 1017 mA h g−1 at the rate of 0.2 C, respectively. After 200 cycles, the PC–K–S electrode retains a reversible capacity of 760 mA h g−1, while only 620 mA h g−1 is left for the PC–U–K–S. Moreover, the columbic efficiency of the PC–K–S cell approaches 99% after several cycles, while the columbic efficiency of PC–U–K–S cells only reaches 93% (see ESI, Fig. S3†). As is well-known, sulfur loading is an important factor that is correlated with the performance. Generally, the lower the sulfur loading, the better the performance of the electrode, however, this is detrimental for practical applications, as high carbon content reduces the energy density per gram of cathode. The battery in our study possessed higher sulfur content than in most previous reports (Table 1), which could be one of the reasons for its a slightly lower capacity compared with those references in which the capacity is calculated based on sulfur only, such as microporous carbon (40% S) with capacity of 1142 mA h g−1,24 and micro–mesoporous carbon sphere (40% S) with capacity of 900 mA h g−1.42 However, if calculation is based on the total mass of composite for carbon–sulfur electrodes, the capacity of our PC–K–S electrodes would be better than the reported data shown in Table 1.
 | ||
| Fig. 6 Discharge–charge voltage profiles of (a) PC–U–K–S and (b) PC–K–S electrodes at various cycles; (c) cycling performances of PC–U–K–S and PC–K–S electrodes at current density of 0.2 C; (d) rate capabilities of PC–U–K–S and PC–K–S electrodes. | ||
| Microporous carbon type | S content (%) | Current rate | Initial capacity (mA h g−1) | Cycle number | Final capacity based on the weight of sulfur (mA h g−1) | Final capacity based on the weight of composite (mA h g−1) |
|---|---|---|---|---|---|---|
| Micro–macroporous carbon [this study] | 68 | 0.2 C | 1017 | 50 | 880 | 598 |
| 100 | 850 | 578 | ||||
| 200 | 760 | 516 | ||||
| MOF-derived microporous carbon21 | 43 | 0.06 C | 1450 | 100 | 490 | 210 |
| Hierarchically porous honeycomb carbon22 | 66.3 | 2 C | 923 | 100 | 564 | 373 |
| Microporous carbon24 | 40 | 0.1 C | 1674 | 200 | 1142 | 456 |
| Hierarchically porous carbon25 | 54 | 0.1 C | 1177 | 50 | 762 | 411 |
| Hierarchically porous carbon monoliths26 | 75 | 0.1 C | 1305 | 25 | 469 | 351 |
| Biomass derived activated carbon27 | 60.1 | 0.2 C | 1258 | 100 | 750 | 450 |
| Micro–mesoporous carbon spheres42 | 40 | 0.1 C | 1550 | 100 | 900 | 360 |
The rate capability of the PC–U–K–S and PC–K–S electrodes is shown in Fig. 6(d). The discharge capacity is decreased with the increase of rate from 0.2 C to 4 C. A satisfactory capacity of 550 mA h g−1 is obtained for the PC–K–S composite electrode, even at 4 C. The rate performance of the PC–K–S electrode is much better than that of the PC–U–K–S electrode, which may be attributed to the facile electronic/ionic transport and enhanced reaction kinetics in the PC–K–S composite.
It is reported that the greater the percentage of micropores in the total pore volume, the better the electrochemical performance that can be obtained.42 In our study, interestingly, the performance of the PC–K–S composite electrode with micro–macroporous structure is much better than that of the PC–U–K–S composite electrode containing only micropores. This contradictory result could be attributed to the unique micro–macroporous interconnected framework of the PC–K carbon matrix. For the PC–U–K with micropores only, a significant amount of sulfur is located on the surface of the PC–U–K–S composite instead of within the pores. It is hard for the micropores to be fully filled with sulfur just by capillary force. The PC–K has a three-dimensional hierarchically porous interconnected structure, however, and the macropores can supply channels for easy sulfur loading into the micropores. This can be concluded from the nitrogen adsorption isotherms of the PC–U–K–S and PC–K–S composites (see ESI, Fig. S4†). The BET surface area for the PC–U–K–S only decreased to 180 m2 g−1, and there still is a peak centered at 0.6 nm, indicating that the micropores were not completely filled with sulfur, so that excess sulfur remained on the surface of the PC–U–K–S composite. However, the specific surface area for the PC–K–S composite significantly dropped to 39 m2 g−1, and the micropores almost disappeared completely, confirming that the storage of sulfur mainly occurs in the micropores. After the introduction of sulfur, the PC–K–S still maintained the original macroporous morphology, and no obvious bulk sulfur could be observed on the surface of the composite in the Fig. 7(a). Based on the observation above, we believe that the macropores play a role in supplying channels for sulfur loading into the micropores instead of encapsulating sulfur. Fig. 7(b) shows the element mapping of sulfur and carbon for the PC–K–S composite and confirms that the sulfur is homogeneously distributed in the framework of the hierarchically porous carbon. In addition, the macroporous structure connected with the micropores in the PC–K–S can facilitate fast transport of electrons/ions and electrolyte, which in turn enhances the electrochemical performance of the sulfur cathode.
 | ||
| Fig. 7 SEM images of PC–K–S composite, and corresponding elemental map images of carbon and sulfur. | ||
In order to further investigate the electrochemical reaction process, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were carried out on the PC–K–S and PC–U–K–S electrodes was carried out (see ESI, Fig. S5 and S6†). CV curves show typical reduction and oxidation peaks representing the reactions of sulfur with lithium during the charge–discharge processes. The sharp redox peaks with stable overlapping features for the PC–K–S electrode confirm the high reversibility and excellent stability of the electrode after first cycle.20,43 In addition, the impedance plots display a semicircular loop standing for the charge-transfer resistance (Rct), which is mainly generated at the interface between the electrode and the electrolyte. Electrolyte resistance Rct of PC–K–S cell remains unchanged even after 200 cycles, while Rct of PC–U–K–S cell increases moderately with the increased cycling. This indicates that the polysulfides could be well confined in the PC–K–S electrode, and thus the dissolution of polysulfides into the electrolyte and the increase in viscosity are successfully avoided. The charge-transfer resistance of PC–K–S is stabilized at around 200 Ω cm2 (calculated after the electrode area normalization) after the first cycle, which suggests a stable electrochemical environment during the cycling process.
4. Conclusion
In summary, a hierarchically porous carbon material with high surface area has been prepared from cotton and used to fabricate a carbon/sulfur composite, which is then explored as cathode material for lithium–sulfur batteries. This unique micro–macroporous structure is found to be directly related to the battery performance. The macroporous structure of this carbon provides channels for sulfur to be loaded into micropores and enhances fast transport of electrons/ions and electrolyte. The microporous structure can trap elemental S and Li polysulfides during cycling. As a result, the composite electrode exhibits an excellent capacity of 760 mA h g−1 after 200 cycles and a good rate capability. More importantly, the hierarchically porous carbon can be mass produced in a simple way at low cost, and the sulfur loading is higher than for most of the reported composites, which makes our S-based electrode highly promising for practical application in lithium–sulfur batteries.Acknowledgements
Financial support provided by the Australian Research Council (ARC) Discovery Project (DP1094261) is gratefully acknowledged. Furthermore, the authors acknowledge Dr Tania Silver for critical reading of the manuscript and valuable remarks.References
- J. Shim, K. A. Striebel and E. J. Cairns, J. Electrochem. Soc., 2002, 149, A1321–A1325 CrossRef CAS PubMed
.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed
- M. K. Song, Y. G. Zhang and E. J. Cairns, Nano Lett., 2013, 13, 5891–5899 CrossRef CAS PubMed
- H. L. Wang, Y. Yang, Y. Y. Liang, J. T. Robinson, Y. G. Li, A. Jackson, Y. Cui and H. J. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS PubMed
- L. C. Yin, J. L. Wang, F. J. Lin, J. Yang and Y. Nuli, Energy Environ. Sci., 2012, 5, 6966–6972 CAS
- P. Novak, K. Muller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–281 CrossRef CAS PubMed
- Y. Yang, G. Y. Zheng, S. Misra, J. Nelson, M. F. Toney and Y. Cui, J. Am. Chem. Soc., 2012, 134, 15387–15394 CrossRef CAS PubMed
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed
- W. Ahn, K. B. Kim, K. N. Jung, K. H. Shin and C. S. Jin, J. Power Sources, 2012, 202, 394–399 CrossRef CAS PubMed
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS PubMed
- G. He, X. L. Ji and L. Nazar, Energy Environ. Sci., 2011, 4, 2878–2883 CAS
- W. Weng, V. G. Pol and K. Amine, Adv. Mater., 2013, 25, 1608–1615 CrossRef CAS PubMed
- X. B. Cheng, J. Q. Huang, H. J. Peng, J. Q. Nie, X. Y. Liu, Q. Zhang and F. Wei, J. Power Sources, 2014, 253, 263–268 CrossRef CAS PubMed
- S. S. Zhang and D. T. Tran, Electrochim. Acta, 2013, 114, 296–302 CrossRef CAS PubMed
- J. T. Lee, Y. Y. Zhao, H. Kim, W. I. Cho and G. Yushin, J. Power Sources, 2014, 248, 752–761 CrossRef CAS PubMed
- N. Azimi, W. Weng, C. Takoudis and Z. C. Zhang, Electrochem. Commun., 2013, 37, 96–99 CrossRef CAS PubMed
- Y. Yang, G. H. Yu, J. J. Cha, H. Wu, M. Vosgueritchian, Y. Yao, Z. N. Bao and Y. Cui, ACS Nano, 2011, 5, 9187–9193 CrossRef CAS PubMed
- F. Wu, J. Z. Chen, L. Li, T. Zhao and R. J. Chen, J. Phys. Chem. C, 2011, 115, 24411–24417 CAS
- Y. Z. Fu and A. Manthiram, J. Phys. Chem. C, 2012, 116, 8910–8915 CAS
- L. F. Xiao, Y. L. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. M. Nie, G. J. Exarhos and J. A. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed
- H. B. Wu, S. Y. Wei, L. Zhang, R. Xu, H. H. Hng and X. W. Lou, Chem.–Eur. J., 2013, 19, 10804–10808 CrossRef CAS PubMed
- Y. H. Qu, Z. A. Zhang, X. H. Zhang, G. D. Ren, X. W. Wang, Y. Q. Lai, Y. X. Liu and J. Li, Electrochim. Acta, 2014, 137, 439–446 CrossRef CAS PubMed
- S. Y. Zheng, Y. Chen, Y. H. Xu, F. Yi, Y. J. Zhu, Y. H. Liu, J. H. Yang and C. S. Wang, ACS Nano, 2013, 7, 10995–11003 CrossRef CAS PubMed
- S. Xin, L. Gu, N. H. Zhao, Y. X. Yin, L. J. Zhou and Y. G. Guo, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef CAS PubMed
- G. Y. Xu, B. Ding, L. F. Shen, P. Nie, J. P. Han and X. G. Zhang, J. Mater. Chem. A, 2013, 1, 4490–4496 CAS
- L. H. Yu, N. Brun, K. Sakaushi, J. Eckert and M. M. Titirici, Carbon, 2013, 61, 245–253 CrossRef CAS PubMed
- J. Zhang, J. Y. Xiang, Z. M. Dong, Y. Liu, Y. S. Wu, C. M. Xu and G. H. Du, Electrochim. Acta, 2014, 116, 146–151 CrossRef CAS PubMed
- M. S. Park, B. O. Jeong, T. J. Kim, S. Kim, K. Y. Kim, J. S. Yu, Y. Jung and Y. J. Kim, Carbon, 2014, 68, 265–272 CrossRef CAS PubMed
- M. Nagao, Y. Imade, H. Narisawa, R. Watanabe, T. Yokoi, T. Tatsumi and R. Kanno, J. Power Sources, 2013, 243, 60–64 CrossRef CAS PubMed
- T. Xu, J. X. Song, M. L. Gordin, H. Sohn, Z. X. Yu, S. R. Chen and D. H. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 11355–11362 CAS
- C. D. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724–4730 CrossRef CAS
- X. Y. Tao, X. R. Chen, Y. Xia, H. Huang, Y. P. Gan, R. Wu, F. Chen and W. K. Zhang, J. Mater. Chem. A, 2013, 1, 3295–3301 CAS
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed
- C. F. Zhang, H. B. Wu, C. Z. Yuan, Z. P. Guo and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 9592–9595 CrossRef CAS PubMed
- B. S. Lee, S. B. Son, K. M. Park, G. Lee, K. H. Oh, S. H. Lee and W. R. Yu, ACS Appl. Mater. Interfaces, 2012, 4, 6701–6709 Search PubMed
- X. Y. Tao, J. T. Zhang, Y. Xia, H. Huang, J. Du, H. Xiao, W. K. Zhang and Y. P. Gan, J. Mater. Chem. A, 2014, 2, 2290–2296 CAS
- S. Dorfler, M. Hagen, H. Althues, J. Tubke, S. Kaskel and M. J. Hoffmann, Chem. Commun., 2012, 48, 4097–4099 RSC
- J. C. Guo, Y. H. Xu and C. S. Wang, Nano Lett., 2011, 11, 4288–4294 CrossRef CAS PubMed
- E. Raymundo-Pinero, P. Azais, T. Cacciaguerra, D. Cazorla-Amoros, A. Linares-Solano and F. Beguin, Carbon, 2005, 43, 786–795 CrossRef CAS PubMed
- C. Lai, X. Gao, B. Zhang, T. Yan and Z. Zhou, J. Phys. Chem. C, 2009, 113, 4712–4716 CAS
- A. T. Ward, J. Phys. Chem., 1968, 72, 4133–4139 CrossRef CAS
- H. Ye, Y. X. Yin, S. Xin and Y. G. Guo, J. Mater. Chem. A, 2013, 1, 6602–6608 CAS
- G. C. Li, G. R. Li, S. H. Ye and X. P. Gao, Adv. Energy Mater., 2012, 2, 1238–1245 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12260g |
| This journal is © The Royal Society of Chemistry 2014 |