
Cu-catalyzed intramolecular hydroarylation of alkynes†
Yun-Long Wang,
Wen-Man Zhang,
Jian-Jun Dai,
Yi-Si Feng* and
Hua-Jian Xu*
School of Chemistry and Chemical Engineering, School of Medical Engineering, Hefei University of Technology, Hefei 230009, P. R. China. E-mail: hjxu@hfut.edu.cn
First published on 11th November 2014
Abstract
An efficient Cu-catalyzed intramolecular hydroarylation reaction of alkynes has been developed. The reaction is accomplished under mild conditions and shows good tolerance to both electron-rich and electron-deficient aryl nucleophiles. A series of aryl, heteroaryl, alkyl, and even N-group attached alkynes are all suitable substrates for the intramolecular hydroarylation.
Introduction
The transition metal-catalyzed hydroarylation of alkynes represents a practical and atom-economical method to form new C(sp2)–C(sp2) bonds via aromatic C–H activation.1,2 In addition, the intramolecular hydroarylation reaction has been recognized as an efficient approach to construct polycyclic aromatic frameworks. Many groups have studied this reaction. For instance, Pd, Pt, Au, Ru, and others precious-metal catalysts catalyzed intramolecular hydroarylation reaction of alkynes have been reported in the past decades.3 Nonetheless, some problems remained to be solved with these reactions. Expensive and/or poisonous catalysts were normally used to promote the reaction in previous studies, which limited the development of large-scale application. The selectivity of exo- and endo-type cyclization products still needs to be controlled. Recently, the Fe-catalyzed intramolecular hydroarylation of alkynes have also been reported,4 but there are still limitations in the types of substrates. For instance, electron-rich aryl nucleophiles are necessary for the intramolecular hydroarylation in Campagne's work.5 On the other hand, the electron-rich aryl nucleophiles underwent the transformation with a very low yield (4-MeO and 2-MeO, 19% and 13%) in Takaki's work.6 Besides, the aryl attachment on the alkyne terminal plays an essential role in above reactions. Therefore, the development of a nontoxic and inexpensive catalyst system with universal functional group compatibility is an interesting challenge.Herein, we describe an general and efficient Cu-catalyzed intramolecular hydroarylation reaction of alkynes under mild conditions (Scheme 1).7 Both of electron-rich and electron-deficient aryl nucleophiles are well tolerated in the reaction to give the 6-endo products in moderate to good yields. Not only the aryl, alkyl, but also the N-group attached alkynes are suitable substrates for the Cu-catalyzed intramolecular hydroarylation. Meanwhile, polycyclic dihydronaphthalenes and chromenes derivatives, which are valuable organic skeleton,8 can be synthesized by present protocol.
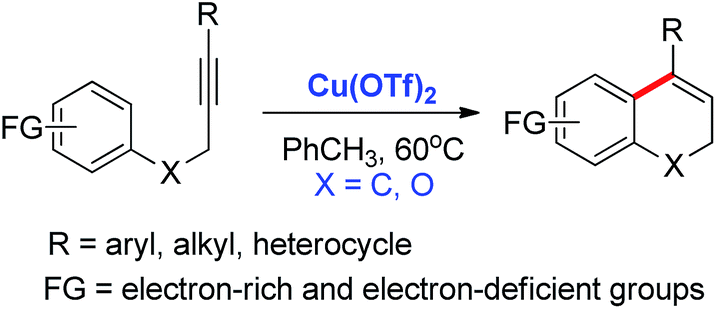 | ||
| Scheme 1 Cu-catalyzed intramolecular hydroarylation reaction of alkynes. | ||
We began our study by examining the intramolecular hydroarylation of 1-methoxy-4-(4-phenylbut-1-ynyl)benzene (I). First, different copper catalysts (entries 1–8) were examined in DCE under 80 °C. As a uniquely effective catalyst, Cu(OTf)2 showed commendable catalytic effect, and the target product was obtained in 22% yield (Table 1, entry 6). Other copper catalysts gave inferior results (Table 1, entries 1–5). In order to improve the product yield, a series of organic solvents were tested (entries 7–13). The mixed solvent of DCE/MeOH showed a better result than either DCE or MeOH (entry 8). When 10 equiv. of MeOH was added in DCE as the reaction solvent, the product yield was improved to 87% (entry 9).9 Surprisingly, PhCH3 showed the best efficiency for this reaction, and the yield of desired product was further increased to 95% (entry 11). In addition, it was our goal to achieve this reaction under milder conditions. It was delighted that when we reduced the temperature to 60 °C, the yield of product was not significantly reduced (entry 13). However, the reaction at 30 °C showed a much low activity under the same optimized conditions (entry 14). Finally, the time of this reaction was also examined (entries 15–17). Gratifyingly, the reaction was completed with an extremely high yield just in 4 hours (entry 17). Disappointedly, no detectable product was obtained when the reaction was underwent in air conditions (entry 18). Furthermore, in the absence of Cu-catalyst, we did not observe the desired product (entry 19).
|
|||||
|---|---|---|---|---|---|
| Entry | CuX | Solvent | T (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: I (0.01 mmol), [Cu] (10 mol%), solvent (1 mL) under Ar atmosphere.b Yield determined by GC.c 10 equiv. of MeOH in DCE.d Isolated yield.e In air. | |||||
| 1 | Cu | DCE | 80 | 12 | NR |
| 2 | CuCl | DCE | 80 | 12 | Trace |
| 3 | CuI | DCE | 80 | 12 | NR |
| 4 | CuCl2 | DCE | 80 | 12 | NR |
| 5 | Cu(OAc)2 | DCE | 80 | 12 | NR |
| 6 | Cu(OTf)2 | DCE | 80 | 12 | 22 |
| 7 | Cu(OTf)2 | MeOH | 80 | 12 | 5 |
| 8 | Cu(OTf)2 | DCE/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
80 | 12 | 48 |
| 9 | Cu(OTf)2 | DCE/MeOHc | 80 | 12 | 87 |
| 10 | Cu(OTf)2 | MeNO2 | 80 | 12 | 18 |
| 11 | Cu(OTf)2 | PhCH3 | 80 | 12 | 95 |
| 12 | Cu(OTf)2 | 1,4-Dioxane | 80 | 12 | Trace |
| 13 | Cu(OTf)2 | PhCH3 | 60 | 12 | 94 |
| 14 | Cu(OTf)2 | PhCH3 | 30 | 12 | 7 |
| 15 | Cu(OTf)2 | PhCH3 | 60 | 1 | 6 |
| 16 | Cu(OTf)2 | PhCH3 | 60 | 3 | 63 |
| 17 | Cu(OTf)2 | PhCH3 | 60 | 4 | 94 (89)d |
| 18e | Cu(OTf)2 | PhCH3 | 60 | 12 | Trace |
| 19 | — | PhCH3 | 60 | 12 | NR |

With the optimized reaction conditions in hand, we next examined the substrate scope of aryl nucleophiles (Table 2). The O-tethered substrate bearing phenyl group afforded the cyclization product in 84% yield (2g). Not only the electron-rich aryl nucleophiles which commonly were used in previous works, were found to undergo the desired reaction in high efficiency, but also the electron-deficient aryl nucleophiles could give the products in moderate to good yields. Many synthetically important functional groups, including ether (2a, 2h), methyl (2b), ketone (2i, 2n), trifluoromethyl (2j, 2m), ester (2k, 2l), lactone (2q), were well-tolerated in the reaction.
|
|---|
| a Reaction conditions: 1 (0.01 mmol), Cu(OTf)2 (10 mol%), PhCH3 (1 mL) at 60 °C for 4 h under Ar atmosphere. Isolated yield, PMP = p-methoxyphenyl.b The solvent is DCE.c DCE. 1 h.d 10 equiv. of MeOH in DCE as the solvent.e DCE, 80 °C.f toluene, 80 °C.g DCE/PhCH3(1:1), 80 °C.h DCE/PhCH3(1:4), 60 °C, 8 h. |
 |
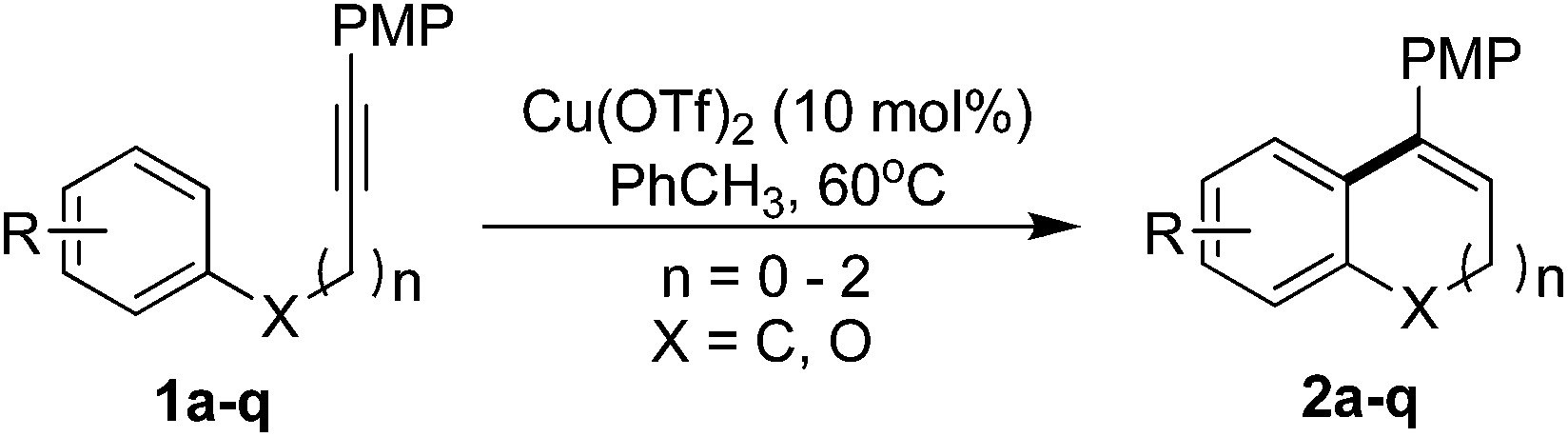
Moreover, arene rings carrying fluoro-, chloro-, and bromo-substituents are compatible with the reaction, enabling additional modifications at the halogenated positions (2c–2f, 2o). Notably, we found that the phenanthrene derivatives can also be achieved in high yield via the cyclization of o-alkynyl biaryls under present reaction conditions (2r). In addition, the successful synthesis of heterocyclic and polycyclic compounds (2p, 2q) made the protocol be potentially used in late-stage intramolecular cyclization. Importantly, the intramolecular hydroarylation of the meta-substituent substrate 1p regioselectively gave the product 2p in 77%. Furthermore, a seven-membered ring can also be formed by this reaction, albeit the yield is relatively lower (2s).10 However, the five-membered ring indene derivative couldn't be formed (2t).
Besides, the effect of terminal attachments R2 in the substituent was investigated (Table 3). In addition to the PMP group, the simple phenyl group as terminal alkyne attachments was also sufficiently reactive, providing a moderate to good yield of the corresponding products (2u, 2y). Compared with the previously developed cationic iron-catalyzed intramolecular hydroarylation alkyne,6 the present method showed a better functional group compatibility, because the substrates with electron-deficient aromatic rings R2 were difficult to obtain the desired products in Takaki's work. However, in our reaction, the R2 group with strong electron-withdrawing CF3 and ester substituted was also well-tolerated and give the product in excellent yield (2v–2x). The substrates with heterocyclic and polycyclic attachments on terminal alkyne, such as thiophene group, oxazolidinone group11 and naphthalene groups, could undergo the hydroarylation reaction to get the product in good yield (2z, 2ab, 2ac). Note that the substrate of oxazolidinone-substituted could be tolerated successfully, which pave a route for the construction of more complex polycyclic aromatic compounds. Surprisingly, alkyne with terminal attachments was also achieved the reaction effectively to obtain the target product in 68% yield (2aa). Yet, there was no reaction occurred when the unsubstituted alkynes was used (2ad).
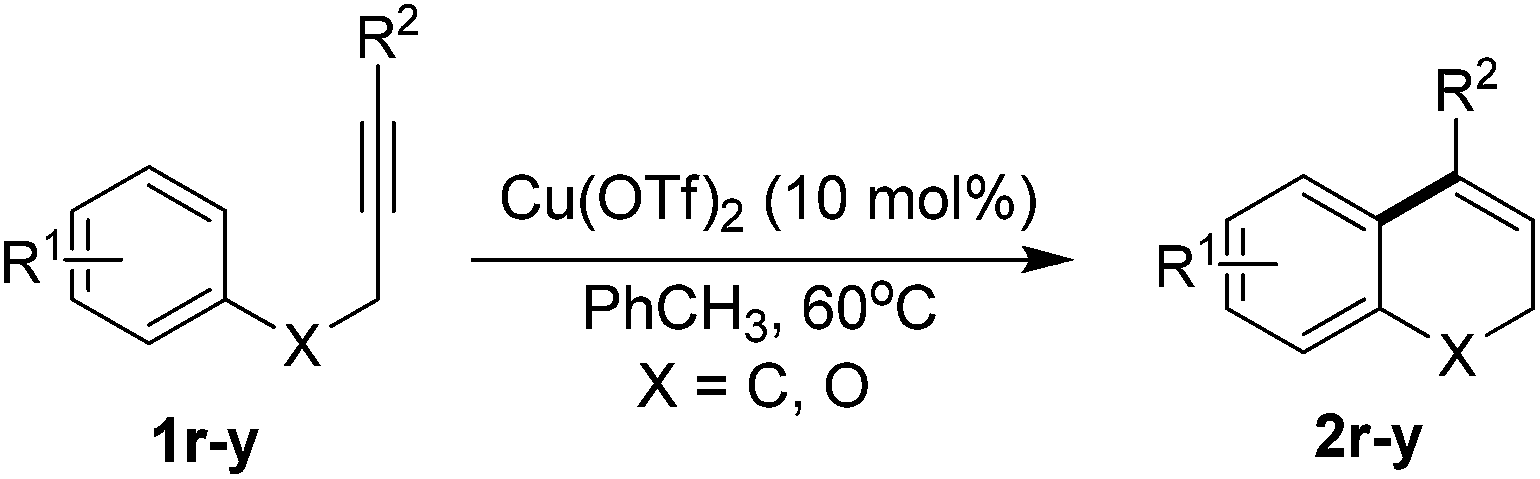

To probe the synthetic utility of this newly developed protocol, a scaled-up reaction was conducted with an aim on evaluating practical aspects. In this experiment, the hydroarylation of 1a was conducted on 1.33 g scale, and product 2a can be obtained in gram quantity with a satisfactory yield of 86% (Scheme 2).12 Notably, a lower loading of Cu catalyst (5 mol%) was used in this gram-scale reaction. Besides, in order to further demonstrate the worth of the products, the further transformation of the model product was also studied (Scheme 3).12 The product II was successfully oxidized by DDQ (1,2-dichloro-4,5-dicyanobenzoquinone) to a polyaromatic naphthalenes III. Then, the hydrogenation of product II can also be occurred smoothly (IV) with good yield. Next, the compound V, which may be an important difluorocyclopropanes derivatives in organofluorine chemistry,13 was obtained by 1,1-difluorocyclopropanation of product II. A series of applications with model product provided a new method to construct the more complicated and valuable polycyclic derivatives.
 | ||
| Scheme 2 Gram-scale reaction. | ||
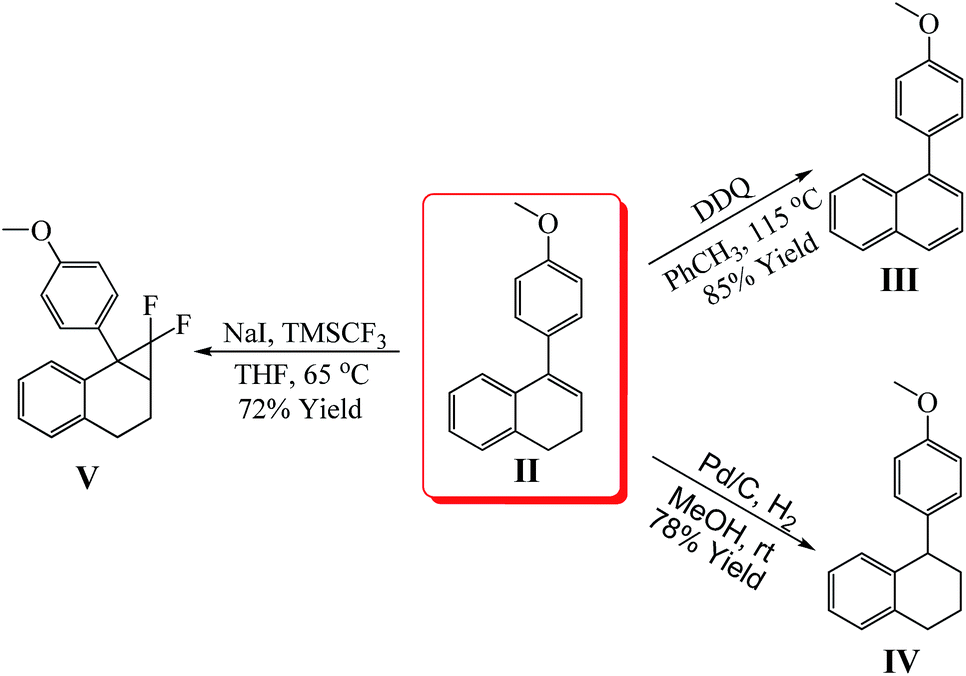 | ||
| Scheme 3 Transformation of model product. | ||
To better understand the reaction mechanism, we performed the kinetic isotope experiment (Scheme 4). The KH/KD values of intramolecular KIE (kinetic isotope experiments) indicated that the C–H cleavage is not the rate-determine step (KH/KD = 0.96). These results suggest that the present Cu-catalyzed cyclization may proceed via a Friedel–Crafts type process.3h,3u,6,14
 | ||
| Scheme 4 Isotope labeling experiment. | ||
Conclusions
In summary, we have reported an efficient Cu-catalyzed intramolecular hydroarylation reaction of alkynes with good functional group tolerance. A nontoxic and inexpensive catalyst, Cu(OTf)2, was successfully used in this hydroarylation reaction, and showed wonderful catalytic effect. Not only the aryl, but also the alkyl and N-group attached alkynes are suitable substrates for this reaction. Numerous 2H-naphthalene and 2H-chromene derivatives can be prepared in good to excellent yields via the straightforward, practically useful method.Experimental
General procedure for the synthesis of the products
A 10 mL over-dried Schlenk tube was charged with corresponding alkyne (0.1 mmol) (if solid), Cu(OTf)2 (10 mol%, 3.62 mg). The tube was evacuated and backfilled with argon (this procedure was repeated three times). Under a counter flow of argon, corresponding alkyne (0.1 mmol) (if liquid) and solvent (1 mL) were added by syringe. The tube was sealed and the mixture was allowed to stir in a preheated oil bath at 60 °C for 4 h. The mixture was cooled to room temperature when the reaction was completed. Next, the organic solvent was concentrated under reduced pressure. The crude product was purified by chromatography on silica gel with petroleum ether/ethyl acetate to give the product 2.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (nos 21272050, 21371044, 21472033) and the Program for New Century Excellent Talents in University of the Chinese Ministry of Education (NCET-11-0627).Notes and references
- For review, see: (a) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS PubMed; (b) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 CrossRef CAS; (c) C. Nevado and A. M. Echavarren, Synthesis, 2005, 167 CAS; (d) M. Bandini, E. Emer, S. Tommasi and A. U. Ronchi, Eur. J. Org. Chem., 2006, 3527 CrossRef CAS; (e) T. Kitamura, Eur. J. Org. Chem., 2009, 1111 CrossRef CAS; (f) X. Wang, L. Zhou and W. Lu, Curr. Org. Chem., 2010, 14, 289 CrossRef CAS; (g) Y. Yamamoto, Chem. Soc. Rev., 2014, 43, 1575 RSC.
- (a) M. Bandini, E. Emer, S. Tommasi and A. U. Ronchi, Eur. J. Org. Chem., 2006, 3527 CrossRef CAS; (b) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2007, 9, 4821 CrossRef CAS PubMed; (c) M. A. Tarselli and M. R. Gagne, J. Org. Chem., 2008, 73, 2439 CrossRef CAS PubMed; (d) H. Harada, R. K. Thalji, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2008, 73, 6772 CrossRef CAS PubMed; (e) T. Kitamura, Eur. J. Org. Chem., 2009, 1111 CrossRef CAS.
- (a) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS; (b) N. Chatani, H. Inoue, T. Ikeda and S. Murai, J. Org. Chem., 2000, 65, 4913 CrossRef CAS PubMed; (c) T. Tsuchimoto, T. Maeda, E. Shirakawa and Y. Kawakami, Chem. Commun., 2000, 1573 RSC; (d) H. Inoue, N. Chatani and S. Muari, J. Org. Chem., 2002, 67, 1414 CrossRef CAS PubMed; (e) A. Fürstner and V. Mamane, J. Org. Chem., 2002, 67, 6264 CrossRef PubMed; (f) M. Nishizawa, H. Takao, V. K. Yadav, H. Imagawa and T. Sugihara, Org. Lett., 2003, 5, 24 Search PubMed; (g) S. J. Pastine, S. W. Youn and D. Sames, Tetrahedron, 2003, 59, 8859 CrossRef CAS PubMed; (h) S. J. Pastine, S. W. Youn and D. Sames, Org. Lett., 2003, 5, 1055 CrossRef CAS PubMed; (i) V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556 CrossRef CAS PubMed; (j) C. E. Song, D. U. Jung, S. Y. Choung, E. J. Roh and S. G. Lee, Angew. Chem., 2004, 116, 6309 (Angew. Chem., Int. Ed., 2004, 43, 6183) CrossRef; (k) C. Nevado and A. M. Echavarren, Chem.–Eur. J., 2005, 11, 3155 CrossRef CAS PubMed; (l) K. S. Kanyiva, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8872 CrossRef CAS PubMed; (m) N. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 5636 CrossRef CAS PubMed; (n) R. Skouta and C. J. Li, Tetrahedron, 2008, 64, 4917 CrossRef CAS PubMed; (o) H. C. Shen, Tetrahedron, 2008, 64, 3885 CrossRef CAS PubMed; (p) R. S. Menon, A. D. Findlay, A. C. Bissember and M. G. Banwell, J. Org. Chem., 2009, 74, 8901 CrossRef CAS PubMed; (q) J. Mo and P. H. Lee, Org. Lett., 2010, 12, 2570 CrossRef CAS PubMed; (r) H. J. Li, R. Guillot and V. Gandon, J. Org. Chem., 2010, 75, 8435 CrossRef CAS PubMed; (s) J. Mo, D. Eom, E. Lee and P. H. Lee, Org. Lett., 2012, 14, 3684 CrossRef CAS PubMed; (t) S. Kim, D. Kang, C. H. Lee and P. H. Lee, J. Org. Chem., 2012, 77, 6530 CrossRef CAS PubMed; (u) A. Arcadi, F. Blesi, S. Cacchi, G. Fabrizi, A. Goggiamanib and F. Marinellia, Org. Biomol. Chem., 2012, 10, 9700 RSC; (v) H. Murase, K. Senda, M. Senoo, T. Hata and H. Urabe, Chem.–Eur. J., 2014, 20, 317 CrossRef CAS PubMed.
- (a) R. Li, S. R. Wang and W. Lu, Org. Lett., 2007, 9, 2219 CrossRef CAS PubMed; (b) T. Hashimoto, T. Izumi, M. S. Kutubi and T. Kitamura, Tetrahedron Lett., 2010, 51, 761 CrossRef CAS PubMed; (c) T. Hashimoto, S. Kutubi, T. Izumi, A. Rahman and T. Kitamura, J. Organomet. Chem., 2011, 696, 99 CrossRef CAS PubMed; (d) D. Eom, J. Mo, Z. M. Gao, S. Kim and P. H. Lee, Eur. J. Org. Chem., 2013, 533 CrossRef CAS.
- C. D. Zotto, J. Wehbe, D. Virieux and J. M. Campagne, Synlett, 2008, 2033 Search PubMed.
- K. Komeyama, R. Igawa and K. Takaki, Chem. Commun., 2010, 46, 1748 RSC.
- Lingaiah and Salunkhe reported Cu/Brønsted acid-catalyzed hydroarylation of alkynes with electron-rich arenes. See: (a) N. Pasha, N. S. Babu, K. T. V. Rao, P. S. S. Prasad and N. Lingaiah, Tetrahedron Lett., 2009, 50, 239 CrossRef CAS PubMed; (b) S. V. Bhilare, N. B. Darvatkar, A. R. Deorukhkar, D. G. Raut, G. K. Trivedi and M. M. Salunkhe, Tetrahedron Lett., 2009, 50, 893 CrossRef CAS PubMed.
- (a) T. Gozler, B. Gozler, A. Patra, J. E. Leet, A. J. Freyer and M. Shamma, Tetrahedron, 1984, 40, 1145 CrossRef CAS; (b) M. A. Castro, M. Gordaliza, J. M. M. Del Corral and A. S. Feliciano, Phytochemistry, 1996, 41, 995 CrossRef CAS; (c) R. S. Ward, Nat. Prod. Rep., 1997, 14, 43 RSC; (d) M. D. Winemiller and W. D. Harman, J. Am. Chem. Soc., 1998, 120, 7835 CrossRef CAS; (e) I. N. Lykakis, C. Efe, C. Gryparis and M. Strakakis, Eur. J. Org. Chem., 2011, 2334 CrossRef CAS.
- Yields of II using various solvents: DCE (1 mL)/MeOH (10 equiv.) (87%) > DCE (1 mL)/MeOH (20 equiv.) (83%) > DCE (1 mL)/MeOH (5 equiv.) (64%) > DCE (1 mL)/MeOH (2 equiv.) (55%).
- The yield of remainder material is 58%.
- 3,4-Dihydronaphthalen-1(2H)-one was got with the yield of 26%, when the reaction was accomplished.
- The specific procedure of experiment. Please see ESI.†.
- (a) F. Wang, T. Luo, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. A. Olah, J. B. Hu and G. K. S. Prakash, Angew. Chem., Int. Ed., 2011, 50, 7153 CrossRef CAS PubMed; (b) L. C. Li, F. Wang, C. F. Ni and J. B. Hu, Angew. Chem., Int. Ed., 2013, 52, 123901 Search PubMed.
- (a) T. J. J. Müller, M. Ansorge and H. J. Lindner, Chem. Ber., 1996, 129, 1433 CrossRef; (b) Y. Kido, S. Yoshimura, M. Yamaguchi and T. Uchimaru, Bull. Chem. Soc. Jpn., 1999, 72, 1445 CrossRef CAS; (c) N. Chatani, H. Inoue, T. Ikeda and S. Murai, J. Org. Chem., 2000, 65, 4913 CrossRef CAS PubMed; (d) T. Tsuchimoto, T. Maeda, E. Shirakawa and Y. Kawakami, Chem. Commun., 2000, 1573 RSC; (e) C. Nevado and A. M. Echavarren, Chem.–Eur. J., 2005, 11, 3155 CrossRef CAS PubMed; (f) E. Soriano and J. M. Contelles, Organometallics, 2006, 25, 4542 CrossRef CAS; (g) M. Y. Yoon, J. H. Kim, D. S. Choi, U. S. Shin, J. Y. Lee and C. E. Song, Adv. Synth. Catal., 2007, 349, 1725 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and 1H and 13C NMR spectra of products. See DOI: 10.1039/c4ra12258e |
| This journal is © The Royal Society of Chemistry 2014 |