
Lamellae evolution of poly(butylene succinate-co-terephthalate) copolymer induced by uniaxial stretching and subsequent heating†
Abstract
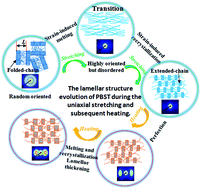
The lamellar structural evolution of biodegradable poly(butylene succinate-co-terephthalate) (PBST) random copolymer was investigated under the conditions of uniaxial stretching at 50 °C, and then heating from 50 to 150 °C at a strain of 150% by the in situ small-angle X-ray scattering (SAXS) technique. A long period referring to the lamellae while processing PBST was calculated, and a schematic for the structural evolution was proposed. It has been found that the lamellar structure experienced a remarkable transformation accompanied by the strain-induced melting of lamellae and formation of new lamellae when the strain exceeded 84% at 50 °C during the initial deformation process. After stretching by 150%, the lamellar structure remained unchanged with the perfection of lamellae in the subsequent heating process from 50 to 120 °C. Only at relatively high temperatures (120–150 °C), the long period of lamellae underwent a significant increase. Conclusions can be drawn that the lamellar structure of PBST is more sensitive to strain and only a relatively high temperature has a prominent impact on it, which is of great significance to provide targeted design guidance for the manufacturing and application of biodegradable PBST copolymer and also gives potential insights into other random and aliphatic–aromatic copolymers.
 Please wait while we load your content...
Please wait while we load your content...

