
Synthesis of novel dispiropyrrolothiazoles by three-component 1,3-dipolar cycloaddition and evaluation of their antimycobacterial activity†
Saoussen Haddadab,
Sarra Boudrigaa,
François Porziob,
Armand Solderab,
Moheddine Askri*a,
Dharmarajan Sriramc,
Perumal Yogeeswaric,
Michael Knorrd,
Yoann Rousseline and
Marek M. Kubickie
aLaboratory of Heterocyclic Chemistry Natural Products and Reactivity/LCHPNR, Department of Chemistry, Faculty of Science of Monastir, 5000 Monastir, Tunisia. E-mail: moheddine.askri@fsm.rnu.tn; Tel: +216 98676187
bDepartment of Chemistry, Quebec Center for Functional Materials, University of Sherbrooke, Sherbrooke, Québec, Canada J1K 2R1
cMedicinal Chemistry and Antimycobacterial Research Laboratory, Pharmacy Group, Birla Institute of Technology & Science-Pilani, Hyderabad Campus, Jawahar Nagar, Hyderabad 500 078, Andhra Pradesh, India
dInstitute UTINAM – UMR CNRS 6213, University of Franche-Comté, 16 Route de Gray, F-25030 Besançon, France
eInstitute of Molecular Chemistry – UMR CNRS 6302, University of Bourgogne, 9 Avenue A. Savary, F-21078 Dijon, France
First published on 3rd November 2014
Abstract
In an on-going effort to develop novel anti-tubercular agents, a series of original dispiropyrrolothiazole derivatives have been synthesized by three-component 1,3-dipolar cycloaddition of (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones, 1,3-thiazolane-4-carboxylic acid and cyclic diketones. The stereochemistry of the spiranic adducts has been confirmed by an X-ray diffraction analysis. Theoretical calculations have been carried out using DFT approach at the B3LYP/6-31G(d,p) level allowing an explanation for the observed regio- and stereoselectivity. The newly synthesized compounds were screened in vitro against Mycobacterium tuberculosis H37Rv and the most active compounds were tested for cytotoxicity studies. Some compounds exhibited significant activity, in particular dispiropyrrolothiazole derivatives 15c and 15f emerged as the most promising antitubercular agents.
Introduction
Tuberculosis (TB), a contagious disease caused mainly by Mycobacterium tuberculosis (MTB), has infected 8.7 million people during 2011 and led to the death of 1.4 million people the same year as reported by the World Health Organization (WHO).1 Moreover, it is estimated that one third of the world's population have latent M. tuberculosis. The spread of this disease was activated by co-infection of patients with the Human Immunodeficiency Virus (HIV).2 The current treatment for susceptible strains prescribed under Directly Observed Treatment Short Course (DOTS) requires at least 6 months of drug therapy using a combination of four drugs that have been discovered in the last four decades.3 However, long duration therapy, associated toxicity and emergence of multi drug-resistant tuberculosis (MDR-TB), highlight the need to explore new potent tuberculosis drugs, exhibiting novel mechanisms of action, with the aim of shortening and improving TB treatment.Nitrogen spiro-heterocycles are important structural building blocks featured in a large number of naturally occurring alkaloids, drugs and pharmaceutically active compounds.4 In particular, spiropyrrolothiazole derivatives are interesting for their exhibition of a wide range of biological activities such as antidiabetic,5 anticancer,6 antimicrobial7 and acetylcholinesterase-inhibitory behaviour.8 It is relevant to point out that spiropyrrolothiazole derivatives bearing oxindolyl 1–5 or acenaphthylenonyl cores 6 also exhibit antimycobacterial properties.9 Many molecules comparable or even better activities than some of the currently employed first-line drugs, ethambutol, isoniazid and pyrazinamide or the second-line agent ciprofloxacin, for TB treatment (Fig. 1).
 | ||
| Fig. 1 Representative antitubercular agents derived from spiropyrrolothiazoles 1–6. | ||
On the other hand, the pyrrolidine-2,5-dione core is found in some alkaloids including Hirsutellone A and B 7 as well as in synthesized compounds 8–9, which have been recognized to display significant growth inhibitory activity against Mycobacterium tuberculosis H37Rv strain (Fig. 2).10
 | ||
| Fig. 2 Examples of N-heterocyclic compounds incorporating a pyrrolidine-2,5-dione motif and exhibiting antimycobacterial activity. | ||
It thus appeared relevant to integrate both the spiropyrrolothiazole and pyrrolidine-2,5-dione motif in one molecule to generate an original scaffold for biological evaluations. The targeted molecules were efficiently synthesized through multi-component sequential reaction of (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones 10, 1,3-thiazolane-4-carboxylic acid 11 and isatin 12 or acenaphthenequinone 13 (Scheme 1). This well reported methodology has already been established as a powerful tool for the construction of spiropyrrolothiazole derivatives using amino acid 11, diketones 12 or 13 and various alkenes. The resulting structures have shown significant interest as an antitubercular precursors.9–11
 | ||
| Scheme 1 Retrosynthesis strategy for synthesis the spiropyrrolothiazole-pyrrolidine-2,5-dione hybrids. | ||
Prasanna and co-workers9c discussed on the first example the different approach mode endo/exo of the possible conformations of azomethine ylide derived from isatin and thiazolidine-4-carboxylic acid toward dipolarophile.
It is worth noting that no theoretical investigation to better understand this kind of reaction have been reported yet. We thus explicitly studied the regio- and stereochemistry from a theoretical point of view by means of the Density Functional Theory (DFT).
Very encouraging activities for some of the synthesized compounds against M. Tuberculosis H37Rv growth are finally reported supporting our approach.
Results and discussion
Synthesis and characterization of the cycloadducts using isatins
The (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones 10 used as starting materials were prepared by Wittig reaction according a protocol described in the literature.12 The reaction of the imides 10 with non-stabilized azomethine ylides, generated in situ by the decarboxylative condensation of 1,3-thiazolane-4-carboxylic acid 11 with various substituted isatin (1H-indole-2,3-dione) derivatives 12 in refluxing methanol, afforded straightforwardly a large series of dispiropyrrolothiazole derivatives 14–16 in a stereoselective manner according Scheme 2. We found no spectroscopic evidence for a competing formation of isomeric compounds 14′–16′.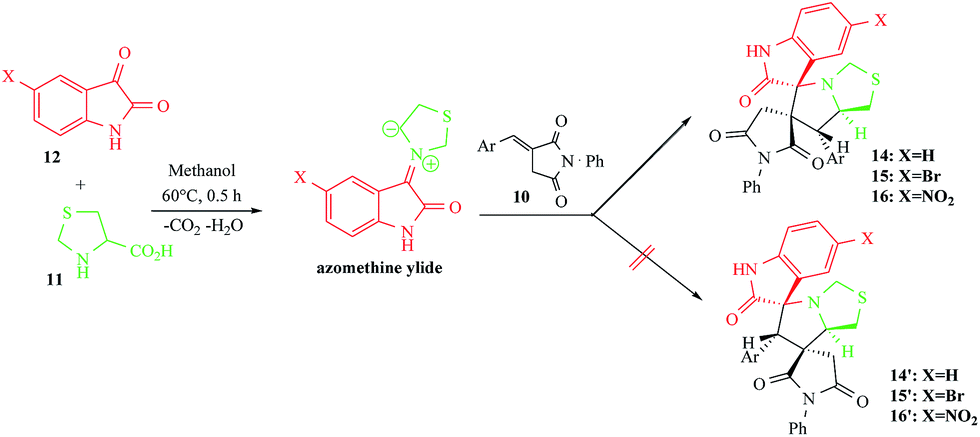 | ||
| Scheme 2 Reaction of (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones 10 with 1,3-thiazolane-4-carboxylic acid 11 and isatin 12. | ||
The targeted spiroadducts 14–16 were obtained as colourless solids in very satisfying yields between 80–92% (Table 1), regardless of the electronic properties of the substituent at the p-position of the aryl group (H, CH3, OCH3, Cl, F and SCH3) of dipolarophile 10.
| Entry | Compound | X | Ar | Yieldb (%) |
|---|---|---|---|---|
| a The reactions were carried out with 10 (1 mmol), 11 (1.5 mmol) and 12 (1 mmol) in methanol (10 mL) at 60 °C for 0.5 h.b Isolated yield after purification by column chromatography.c Failure to separate. | ||||
| 1 | 14a | H | C6H5 | 89 |
| 2 | 14b | H | p-MeC6H4 | 83 |
| 3 | 14c | H | p-MeOC6H4 | 86 |
| 4 | 14d | H | p-ClC6H4 | 91 |
| 5 | 14e | H | p-FC6H4 | 80 |
| 6 | 14f | H | p-CH3SC6H4 | 83 |
| 7 | 15a | Br | C6H5 | 87 |
| 8 | 15b | Br | p-MeC6H4 | 91 |
| 9 | 15c | Br | p-MeOC6H4 | 83 |
| 10 | 15d | Br | p-ClC6H4 | 89 |
| 11 | 15e | Br | p-FC6H4 | 90 |
| 12 | 15f | Br | p-CH3SC6H4 | 82 |
| 13 | 16a | NO2 | C6H5 | 80 |
| 14 | 16b | NO2 | p-MeC6H4 | 86 |
| 15 | 16c | NO2 | p-MeOC6H4 | 88 |
| 16 | 16dc | NO2 | p-ClC6H4 | — |
| 17 | 16e | NO2 | p-FC6H4 | 87 |
| 18 | 16f | NO2 | p-CH3SC6H4 | 85 |
The structure of the spiroadducts was characterized by IR, 1H NMR, 13C NMR and an X-ray structure analysis, as exemplified for cycloadduct 14a. The IR spectrum of 14a exhibits absorptions at 1712, 1774 and 3166 cm−1 due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxindole, CO imide and N–H stretching vibrations, respectively. Selected 1H and 13C chemical shifts assignment of 14a are shown in Fig. 3.
O oxindole, CO imide and N–H stretching vibrations, respectively. Selected 1H and 13C chemical shifts assignment of 14a are shown in Fig. 3.
 | ||
| Fig. 3 Selected 1H and 13C NMR chemical shifts of 14a. | ||
The 1H NMR spectrum of 14a exhibits two doublets at δ 2.67 and δ 3.61 ppm (J = 18.3 Hz) corresponding to the 4′′-CH2 group. The hydrogen atoms from 1-CH2 appear as a doublet of doublets at 2.98 (J = 10.5 and 4.8 Hz) and 3.15 ppm (J = 10.5 and 6.0 Hz). The doublets at 3.74 and 3.88 ppm (J = 8.2 Hz) are assigned to the 3-CH2 hydrogen atoms. The H-7 and H-7a protons appear as a doublet at 4.25 ppm (J = 9.9 Hz) and a multiplet at 4.79–4.86 ppm, respectively. The multiplicity of the signals, doublet and multiplet, are clearly corroborating the regiochemistry of the cycloaddition reaction. If the hypothetical alternative regioisomer 14′a (Scheme 2) would have been formed, the pyrrolizidinyl protons H-7 and H-7a should give rise to a singlet and doublet of doublet pattern in the 1H NMR spectrum. A board singlet broad at 8.11 ppm indicates the presence of the NH proton of the oxindole ring.
The 13C NMR spectrum shows two peaks at δ 65.0 and 75.9 ppm, corresponding to the two spirocarbons C-6 and C-5. The occurrence of peaks at δ 173.1, 175.5 and 177.6 ppm confirm the presence of three carbonyl groups. In the DEPT-135 spectrum, the peaks at δ 34.5, 34.8 and 50.7 ppm indicate the presence of three methylene groups. The pyrrolizidinyl carbons C-7 and C-7a appear at δ 53.1 and 70.4 ppm, respectively. The reactions were found to be highly regioselective leading to the formation of only one isomer 14a, no indication for the presence of regioisomer 14′a was found (Scheme 2).
The stereochemical outcome of the cycloaddition was also corroborated by an X-ray diffraction study of the crystal structure of the cycloadduct 14a, whose ORTEP diagram is displayed in Fig. 4. The molecules of 14a crystallize in the centro-symmetric triclinic space group P-1. Consequently, one intrinsically chiral molecule (as the one shown in Fig. 4) with C9, C11, C21 and C22 atoms in R chirality has an opposite isomer (enantiomer) giving rise to the overall racemic structure.
 | ||
| Fig. 4 ORTEP of the molecular structure of cycloadduct 14a in the crystal at 50% probability level (115 K). For clarity, only stereo-chemically significant hydrogen atoms are shown. Selected bond lengths (Å) and angles (°): C9–C8 1.5345(16), C9–C10 1.5256(16), C9–C11 1.5799(17), C9–C22 1.5515(16), C11–N2 1.4695(15), C11–C12 1.5644(16), C11–C14 1.5318(17); C7–N1–C10 111.95(10), C19–N2–C21 111.25(10), C19–N2–C11 117.42(10), C11–N2–C21 110.45(9), C12–N3–C13 111.46(10). | ||
The fused heterocycle formed by 9 atoms (C11, C12, N3, C13 through C18) is almost planar with the largest deviation from the best least square plane of some 0.06 Å observed for the C11 and C12 atoms. This suggests a high degree of π conjugation in this part of the molecule. The dihedral angles including both spiranic carbon atoms C9 and C11 are close to 90°: 85.66(7)° for the C11/C12/C14//N2/C11/C9 and 89.45(7)° for the C11/C9/C22//C8/C9/C10 planes. One also notes that the N1 and N3 atoms are in a perfect trigonal planar hybridization (sum of three angles around are equal to 359.8 and 360°, respectively). The N2 atom adopts a pyramidal structure with the sum of three angles between the σ bonds in order of 339.1°.
The ORTEP diagram reveals that (i) the two carbonyl carbons C-2′′ (C-10 in ORTEP) and C-2′(C-12 in ORTEP) are in trans-relationship, and (ii) reveals a cis-relationship between the proton attached at C-7 (C-22 in ORTEP) and the carbonyl carbon C-2′.
Thus, the cycloadduct is formed through an exo-approach between the (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones and anti-12a (Scheme 2). The cycloaddition proceeded via an exo-transition state affording only one diastereomer, as found in many similar cycloaddition studies.9–11
DFT calculations
To better understand the observed regio- and stereoselectivity, the 1,3-dipolar cycloaddition reactions of the azomethine ylide 12a with dipolarophile 10a has been theoretically studied using the DFT approach. Such calculations aim to unveil the reaction pathway predicted by DFT to shed light on the experimental behaviour. All the Gibbs energies that are discussed in the text have been computed within the harmonic approximation at a temperature of 298.15 K, and a pressure of 1.0 atmosphere and within the harmonic approximation. The optimized geometry of the dipole 12a reveals its planar structure; it actually exists in two diasteromeric syn- and anti-forms (Scheme 3). The syn-12a form is slightly more stable than the anti-12a form with a difference between their respective Gibbs energy of ΔGsyn–anti = 0.40 kcal mol−1. This energy difference is lower that the thermal energy at 298 K (kBT = 0.6 kcal mol−1). The attack of the dipolarophile on syn-12a results in an unfavourable inward movement of the 1,3-thiazolane ring towards the isatin ring, which lead to the steric hindrance between these later nucleus. While on anti-12a, it results in a favourable outward movement13 (Scheme 3). Hence, the computational investigation is focused on the anti-12a conformation. | ||
| Scheme 3 Gibbs energies of anti- and syn- forms of azomethine ylide 12a calculated at the B3LYP/6-31G(d,p) level. The mode of the attack of the dipolarophile is also represented. | ||
A study using the frontier molecular orbital (FMO) model14 was first carried out. This theory is based on the statement that a valuable estimate of the actual reactivity can be achieved by unveiling interaction between the HOMO and the LUMO of the reactants.15 As shown in Fig. 5, the energy gap HOMOanti-12a–LUMO10a (3.817 eV) is lower than the LUMOanti-12a and the HOMO10a (4.949 eV). This attests that the HOMOdipole–LUMOdipolarophile interaction controls the cycloaddition reaction within a normal electron demand reaction.
 | ||
| Fig. 5 HOMO and LUMO energies of anti-12a and dipolarophile 10a calculated at the B3LYP/6-31G(d,p) level. | ||
The analysis of the global and local indices for electrophilic/nucleophilic is a powerful tool to grasp the reactivity in polar cycloadditions. In Table S1 (in the ESI†), the static global properties, namely electronic chemical potential μ, chemical hardness η, and the electrophilicity index ω are reported. We can notice that μ of dipole-1,3 (−3.776 eV) is greater than μ of dipolarophile 10a (−4343 eV) and η of anti-12a (4.170 eV) is lower than η of 10a (4596 eV). Consequently, the charge transfer will take place from ylide anti-12a to dipolarophile 10a. On the other hand, the electrophilicity values show that ω of dipolarophile 10a (2052 eV) is greater than ω of azomethine ylide anti-12a (1.710 eV), indicating that anti-12a will act as a nucleophile whereas 10a will act as an electrophile.
After evaluating the global nucleophilic/electrophilic character, a local reactivity is analyzed by means of the condensed Fukui functions, the cationic and anionic systems were kept at the optimized geometries of the corresponding neutral systems. Fukui function parameters were evaluated by using the technique of electrostatic potential (ESP) derived atomic populations. The calculated local chemical reactivity parameters of anti-12a and dipolarophile 10a are shown in Table S2 (in the ESI†). In the reaction between dipolarophile 10a and azomethine ylide anti-12a, the most favored interactions take place between the C-5′′ centre of anti-12a (possessing the highest value of f− = 0.253) and the C-6 center of 10a (possessing the highest value of f+ = 0.154). These results are in prefect agreement with the afore-mentioned experiment (Scheme 2).
The four reactions shown in Scheme 4 were theoretically investigated. They consist of (i) two regioisomeric channels and (ii) endo and exo stereoisomeric approach modes between the anti-dipole 12a and the dipolarophile 10a. The cycloadducts were labelled endo-14a, exo-14a, endo-14′a, exo-14′a and their transition states (TS) endo-TS1, exo-TS1, endo-TS2, exo-TS2 were energetically optimized and characterized under the same level of calculation by using the Berny analytical gradient method.16 These TS, which are characterized by saddle point leading to the occurrence of one imaginary frequency, are displayed in Fig. 6.
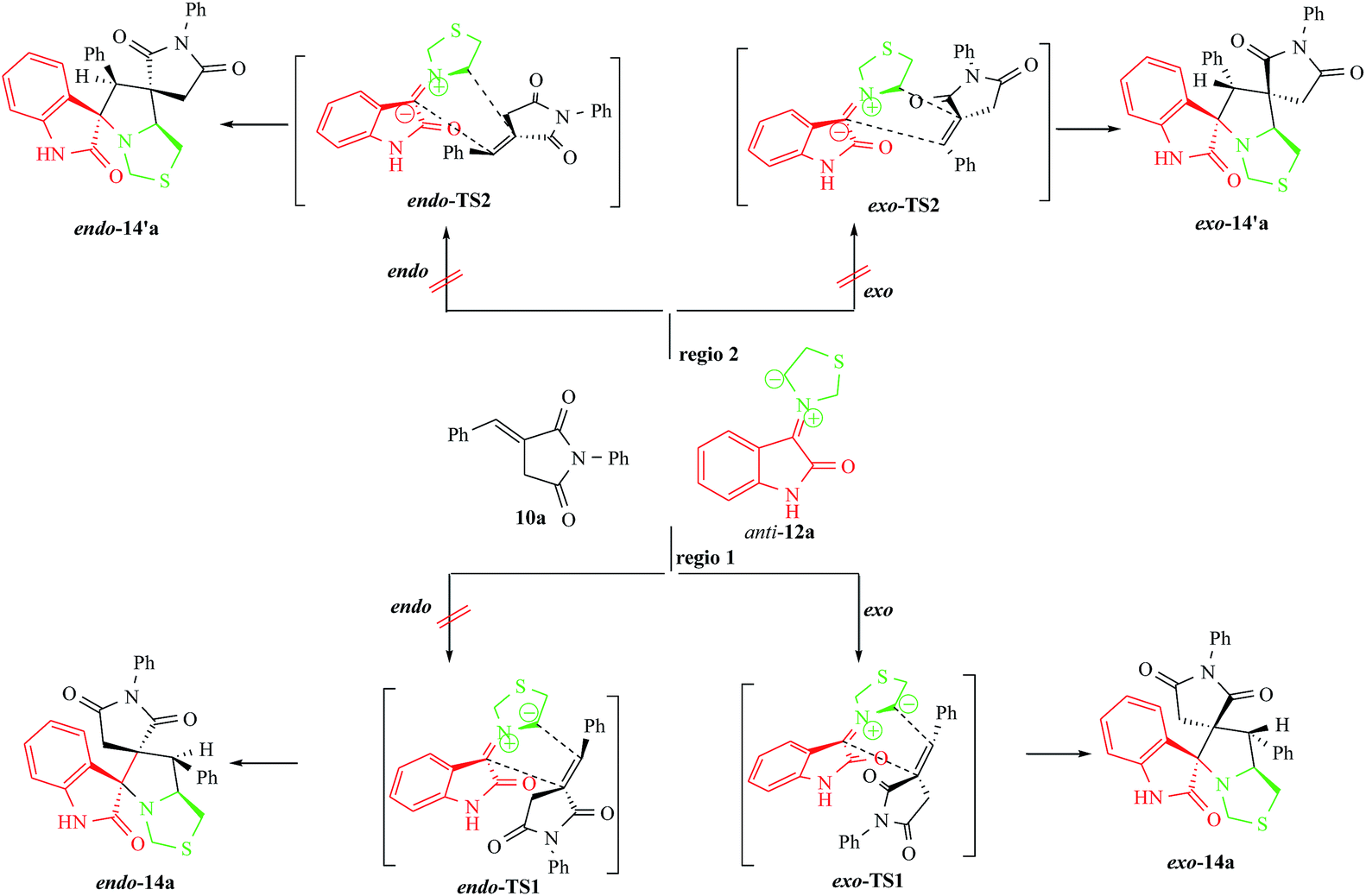 | ||
| Scheme 4 Plausible mechanism for the formation of 14a. | ||
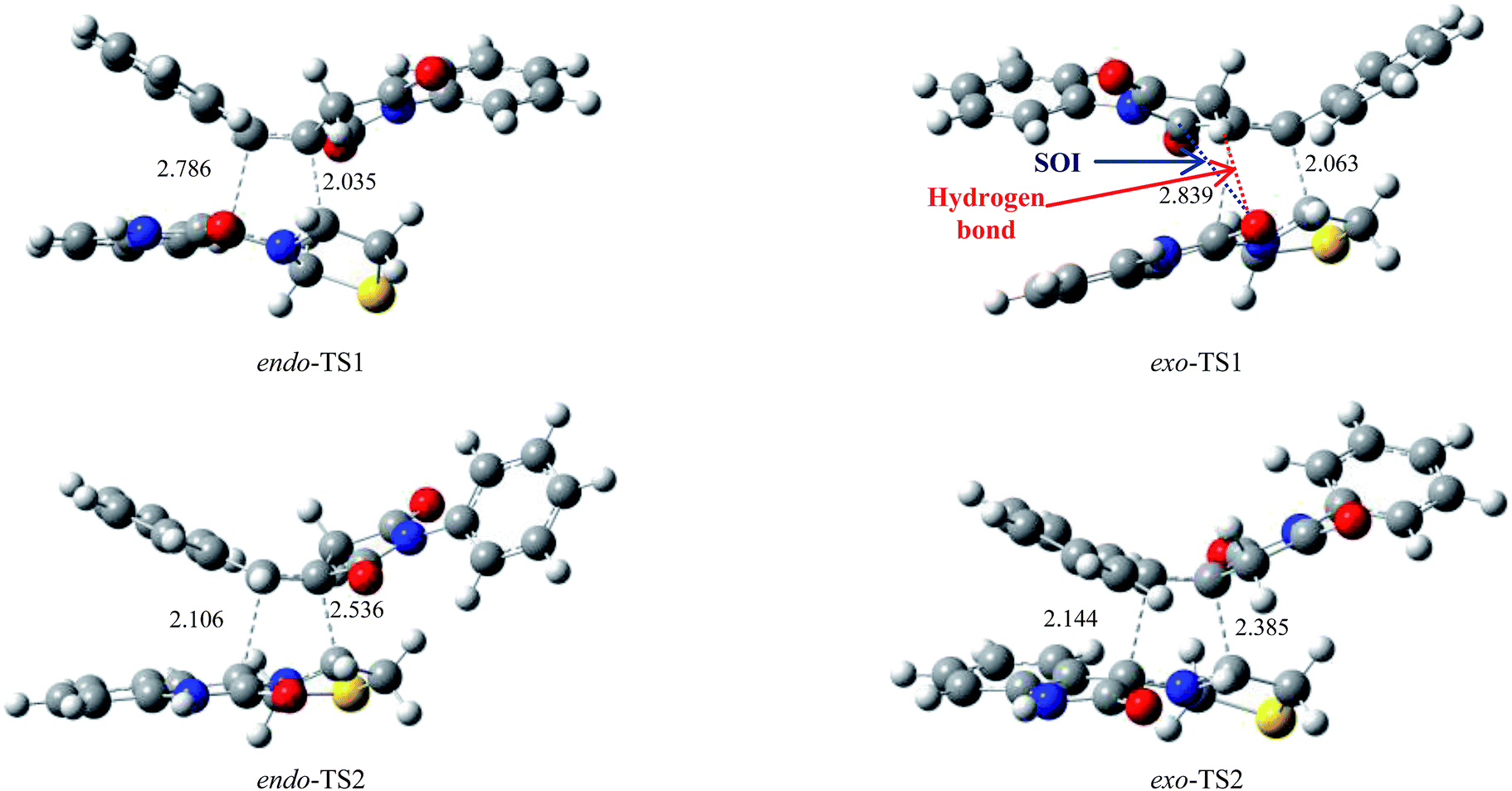 | ||
| Fig. 6 The four possible transition states for the 1,3-dipolar cycloaddition of anti-12a across 10a optimized at the B3LYP/6-31G(d,p) level. The values of the lengths of the C–C bonds directly involved in the reaction are given in angstroms. Coordinates are given in the ESI.† | ||
At the TSs associated with the regioisomeric channels 1, the length of the C-3–C-3′ forming bonds are 2.786 Å at endo-TS1 and 2.839 Å at exo-TS1, while the distance between the C-6 and the C-5′′ atoms is 2.035 Å at endo-TS1 and 2.063 Å at exo-TS1. Considering the TSs associated with the regioisomeric channels 2, the length of the C-6–C-3′ forming bonds are 2.536 Å at endo-TS2 and 2.385 Å at exo-TS2. The extent of the asynchronicity on the bond-formation can be measured by means of the difference between the lengths of the two σ bonds that are being formed in the reaction that is, Δd = d1 − d2.16 asynchronicity at the regioisomeric channels 1 is Δd = 0.750 Å at endo-TS1 and 0.776 Å at exo-TS1, while that at the regioisomeric channels 2 is Δd = 0.430 Å at endo-TS2 and 0.241 Å at exo-TS2. These geometrical parameters show that these cycloadditions correspond to asynchronous concerted processes. Moreover, TSs corresponding to the regioisomeric channels 1 are more advanced.An analysis of the IRC17 cycloaddition profile, using the Hessian based predictor–corrector (HPC) method18 were used to confirm that these 1,3-dipolar cycloaddition reactions take place along asynchronous concerted processes. These results are in agreement with previous studies.19
The Gibbs free energy (ΔG), enthalpy (ΔH) and entropy (ΔS), for the different stationary points along the reactive channels are reported in Table 2. All the energy values have been corrected for zero point energy. The kinetic parameters (activation energy, activation enthalpies and activation entropies, between reactants and transition states) are first discussed. It is observed that the activation energy for the TSs increases in the following order: exo-TS1 < exo-TS2 < endo-TS1 < endo-TS2. The most favourable pathway, via the exo-TS1, exhibits a very lower activation energy (20.3 kcal mol−1) than that stemmed from the second most probable route (26.3 kcal mol−1). The exo-TS1 is thus the kinetically favourable pathway. Investigation of activation enthalpies and entropies for these cycloadditions also revealed that the exo-TS1 pathway is more favourable from these two viewpoints. From an energy point of view, structural investigation (Fig. 6) of these two TS reveals that the dipole and dipolarophile are largely superimposed in the exo-TS1. Thus, the significantly lowest energy value of exo-TS1 can be partially explained by secondary orbital interactions (SOI) in exo-TS1, which occurs between the oxygen atom of the carbonyl of the isatin and the carbon atom of the carbonyl of the dipolarophile. Moreover, an additional hydrogen bond takes place through in exo-TS1, between one of the methylene hydrogen atoms of dipolarophile 10a with the carbonyl of the azomethine ylide 12a (2.406 Å).20
| ΔG | ΔH | ΔS | |
|---|---|---|---|
| endo-TS1 | 28.9 | 14.4 | −48.7 |
| exo-TS1 | 20.3 | 4.9 | −51.6 |
| endo-TS2 | 30.7 | 16.5 | −47.5 |
| exo-TS2 | 26.3 | 11.3 | −50.3 |
| endo-14a | 3.0 | −13.0 | −53.9 |
| exo-14a | −1.4 | −17.4 | −53.4 |
| endo-14′a | −1.8 | −17.3 | −52.1 |
| exo-14′a | −0.7 | −16.6 | −53.2 |
The analysis of the thermodynamic parameters (Gibbs free energy (ΔG), enthalpy (ΔH) and entropy (ΔS), between reactants and products) of the studied reactions. It is observed that the lowest ΔG values stem from the endo approach on 14′a. However, the Gibbs free energy difference between the two products of lowest energies (endo-14′a and exo-14a, −1.8 and −1.4 kcal mol−1, respectively) is underneath the thermal energy value at 298.15 K (0.6 kcal mol−1). Moreover, all cycloadditions are exothermic processes, and ΔH is in the range of −13.0 to −17.4 kcal mol−1. Contrary to kinetic parameters, it appeared that both entropies and enthalpies are similar among the most favorable thermodynamics products. This evidence thus suggests that a kinetic control takes place. Accordingly, DFT investigation shows that the most favorable pathways goes through exo-TS1 (exo-14a) under kinetic control. Such a conclusion is in perfect agreement with the experimental observation assigning the exo-regioisomer 14a as the only formed product.
Synthesis and characterization of the cycloadducts using acenaphthenequinone
To investigate further the regio- and stereoselectivity of the cycloaddition, we carried out the three-component 1,3-dipolar cycloaddition reaction of the dipolarophile 10 with another azomethine ylide generated in situ from 1,3-thiazolane-4-carboxylic acid 11 and acenaphthenequinone 13 as diketones. Under similar reaction conditions, this reaction afforded the corresponding dispiropyrrolothiazole derivatives 17a–e in good yields (82–90%) as shown in Scheme 5 and Table 3. | ||
| Scheme 5 Reaction of (E)-3-arylidene-1-phenyl-pyrrolidine-2,5-diones 10 with 1,3-thiazolane-4-carboxylic acid 11 and acenaphthenequinone 13. | ||
The structural and stereochemical features of all derivatives in series 17 are fully supported by the IR and NMR spectroscopic data. As illustrated for compound 17b, two intense IR absorption bands at 1709 and 1791 cm−1, are observed in the IR spectrum. They correspond to the acenaphtenone and imide ring carbonyls, respectively. The 1H and 13C spectroscopic data of 17b are summarized in Fig. 7.
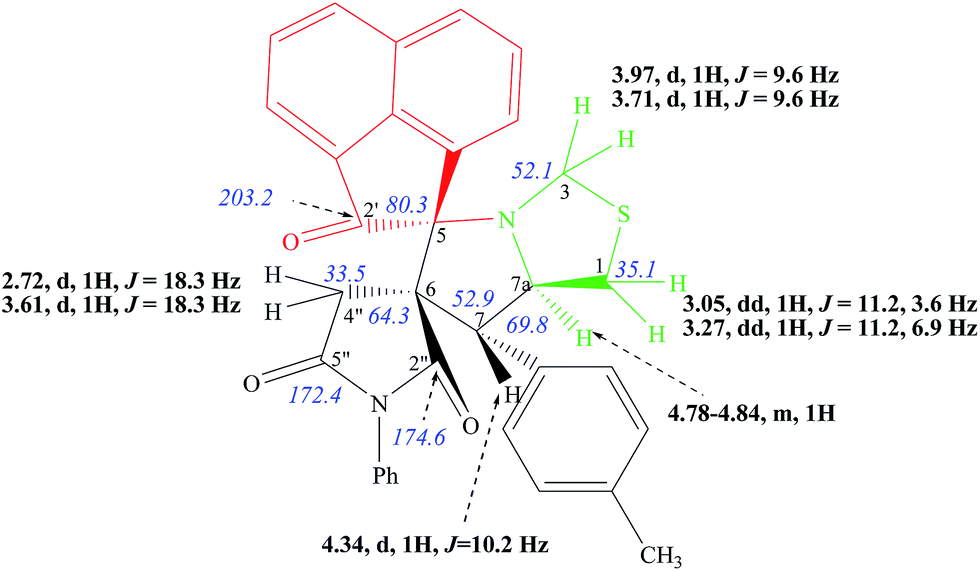 | ||
| Fig. 7 Selected 1H and 13C NMR chemical shifts of 17b. | ||
The 1H NMR spectrum of 17b displays a doublet at δ 4.34 ppm (J = 10.2 Hz) and a multiplet between 4.78–4.84 ppm, corresponding to the H-7 and H-7a protons, respectively. These data confirm the regiochemistry of the cycloaddition reaction. The signals at 64.3 ppm and 80.3 ppm in the 13C NMR spectrum of 17b are attributed to the presence of two spiro-carbons C-6 and C-5, respectively. The resonance at 203.2 ppm is characteristic of the presence of the carbonyl group from the acenaphthenequinone moiety.
Biological activities
In vitro MTB screening
All new compounds were screened for their in vitro anti-mycobacterial activity against M. tuberculosis H37Rv using the agar dilution method21 and drug concentrations from 50 μg mL−1 to 0.78 μg mL−1. The MIC is defined as the minimum concentration of compound required to achieve complete inhibition of bacterial growth. This method agrees with the procedure recommended by the National Committee for Clinical Laboratory Standards for the determination of MIC in triplicate. The MIC's of the synthesized compounds 14–17 are reported in Table 4 along with the MIC's of several standard drugs for comparison.| Entry | Comp. | X | Ar | MTBa (MIC) μg mL−1 | (RAW 264.7 cells) % inhibition |
|---|---|---|---|---|---|
| a Mycobacterium tuberculosis H37Rv. NT: not tested. ND: not determined. | |||||
| 1 | 14a | H | C6H5 | 3.125 | 32.4 |
| 2 | 14b | H | p-MeC6H4 | 25 | ND |
| 3 | 14c | H | p-MeOC6H4 | 3.125 | 28.63 |
| 4 | 14d | H | p-ClC6H4 | 25 | ND |
| 5 | 14e | H | p-FC6H4 | 25 | ND |
| 6 | 14f | H | p-CH3SC6H4 | 50 | ND |
| 7 | 15a | Br | C6H5 | NT | NT |
| 8 | 15b | Br | p-MeC6H4 | 50 | ND |
| 9 | 15c | Br | p-MeOC6H4 | 1.56 | 27.63 |
| 10 | 15d | Br | p-ClC6H4 | 6.25 | 30.16 |
| 11 | 15e | Br | p-FC6H4 | 12.5 | ND |
| 12 | 15f | Br | p-CH3SC6H4 | 1.56 | 20.74 |
| 13 | 16a | NO2 | C6H5 | NT | NT |
| 14 | 16b | NO2 | p-MeC6H4 | 6.25 | 28.15 |
| 15 | 16c | NO2 | p-MeOC6H4 | 12.5 | ND |
| 16 | 16d | NO2 | p-ClC6H4 | NT | NT |
| 17 | 16e | NO2 | p-FC6H4 | 25 | ND |
| 18 | 16f | NO2 | p-CH3SC6H4 | 50 | ND |
| 19 | 17a | H | C6H5 | 3.125 | 35.62 |
| 20 | 17b | H | p-MeC6H4 | 50 | ND |
| 21 | 17c | H | p-MeOC6H4 | 3.125 | 43.12 |
| 22 | 17d | H | p-ClC6H4 | 25 | ND |
| 23 | 17e | H | p-FC6H4 | 25 | ND |
| Isoniazid | 0.05 | ||||
| Rifampicin | 0.1 | ||||
| Ciprofloxacin | 3.13 | ||||
| Ethambutol | 1.56 | ||||
| Pyrazinamide | 6.25 | ||||
The spiroheterocycles 14–17 have MICs in the range of 1.56–50 μg mL−1 (Table 3). Among them, eight compounds (14a, 14c, 15c, 15d, 15f, 16b, 17a and 17c) were found to be more or equally active against MTB than the first line anti-TB drug pyrazinamide (MIC 6.25 μg mL−1), whilst six of them 14a, 14c, 15c, 15f, 17a and 17c were more potent than ciprofloxacin (MIC 3.13 μg mL−1). All compounds are however less potent than rifampicin and isoniazid. The dispiropyrrolothiazole derivatives 15c and 15f displayed the maximum potency with MICs of 1.56 μg mL−1, thus being equipotent with ethambutol. Noteworthy is the fact that the latter two heterocycles are two and four times more potent than the standard drugs ciprofloxacin and pyrazinamide, respectively.
The influence of the substituents at the isatin core as well as on the aryl rings was examined for structure–activity relationship (SAR). As shown in Table 3, the substituent present on the isatin nucleus has a very strong effect on the activity of spiropyrrolothiazole derivatives 14–16. The order of activity, in general, being Br > H > NO2 was evidenced from the observation that three compounds in series 15 (15c, 15d and 15f), two in series 14 (14a and 14c), and one in series 16 (16b) were more active against MTB (Table 3).
In vitro cytotoxicity screening
Some of the selected compounds were also tested for in vitro cytotoxicity against RAW 264.7 cells at 50 μM concentration using a (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The percentage of cells inhibition is reported in Table 3. The most promising anti-TB compounds 15c and 15f showed 27.63 and 20.74% inhibition, respectively, at 50 μM.Conclusions
In this study we demonstrated that the literature-known three-component 1,3-dipolar cycloaddition of azomethine ylides generated in situ can also be applied to the synthesis of a series of novel dispiropyrrolothiazole derivatives with isatin or acenaphthenequinone using arylidene-succinimide as alkene component. This reaction can be conducted under convenient and mild reaction conditions, affording efficiently the cycloaddition products with high yields in a complete regio- and stereoselective manner.An analysis based on theoretical calculations using the DFT approach, B3LYP/6-31G(d,p) showed that the spirocycloadduct 14 is obtained through a 1,3-dipolar cycloaddition reaction via a high asynchronous mechanism with a very low activation energy, as compared to the three other possible reaction paths. This outcome is in agreement with the experimental observations.
Screening all these derivatives against Mycobacterium tuberculosis H37Rv and cytotoxicity revealed that 15c and 15f are the most active antitubercular agents compared to the other evaluated compounds.
Experimental
General information and apparatus
NMR spectra were recorded with a Bruker-Spectrospin AC 300 spectrometer operating at 300 MHz for 1H and 75 MHz for 13C using tetramethylsilane (TMS) as internal standard (0.00 ppm) in CDCl3 as solvent. The following abbreviations were used to explain the multiplicities: bs = broad singlet, s = singlet, d = doublet, dd = doublet of doublets, m = multiplet. IR spectra were recorded on a Perkin-Elmer Spectrum Two™ FT-IR in the ATR mode. Elemental analyses were performed on a Perkin Elmer 2400 Series II Elemental CHNS analyzer. Materials: thin-layer chromatography (TLC): TLC plates (Merck, silica gel 60 F254 0.2 mm 200 × 200 nm); substances were detected using UV light at 254 nm.General procedure for the preparation of cycloadducts 14–17
A mixture of 10 (1.0 mmol), 1,3-thiazolane-4-carboxylic acid 11 (1.5 mmol) and isatin 12 or acenaphthenequinone 13 was refluxed in methanol (10 mL) for 0.5–1 h. After completion of the reaction as monitored from TLC, the mixture was poured into ice-water (50 mL). The resulting solid was filtered off and purified by flash column chromatography on silica gel employing ethylacetate-cyclohexane (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 v/v) as eluent to obtain the pure products 14–17. Spectroscopic data for the all compound are presented in the ESI.†
:7 v/v) as eluent to obtain the pure products 14–17. Spectroscopic data for the all compound are presented in the ESI.†
DFT calculations
All calculations have been performed using the Gaussian 09 package.22 The hybrid density functional theory B3LYP23 with the 6-31G(d,p) basis set24 was employed to compute the energy. The optimization procedure was carried out using the Berny analytical gradient method.16 The stationary points were characterized through frequency calculations in order to ensure that the minima in energy, i.e. reactants and products, as well as transition states (TS) exhibit zero and one imaginary frequency, respectively. Intrinsic reaction coordinate (IRC)17 calculations starting at the saddle points were carried out to check the connections between the TS, reactants and cycloadducts, using the Hessian based predictor–corrector (HPC) method.18X-ray structure study
A colourless single-crystal of 14a has been mounted on a Nonius Kappa Apex II diffractometer and the intensity data have been collected at 115 K using MoKα radiation of λ(Mo-Kα) = 0.71073 Å. These data were further treated with the SAINT V8.27B program suite (Bruker AXS Inc., 2012) within the OLEX2 frame.25 The model of the structure has been solved by direct methods with SHELXS-97 and refined with SHELXL-97.26Crystal and refinement data for 14a: C28H23N3O3S; M = 481.55; crystal system triclinic; space group P-1, a = 9.1367(9) Å, b = 10.0617(10) Å, c = 14.3025(15) Å, α = 94.058(4), β = 99.914(4), γ = 114.73(4)°, V = 1160.7(2) Å3, Z = 2, λ(Mo-Kα) = 0.71073 Å, F(000) = 504, μ(Mo-Kα) = 0.082 mm−1, T = 115(2) K. 44058 reflections collected, 5333 unique and 4569 with I > 2σ(I). Final agreement factors: R1 = 0.0418 (all observed) and 0.0336 with I > 2σ(I), wR2 = 0.0888 (all observed) and 0.0832 with I > 2σ(I). GOF = 1.015. Final residuals ρmax = 0.371, ρmin = −0.304 e− Å−3.†
Biological evaluation
In vitro MTB screening
Two-fold serial dilutions of each test compound/drug were prepared and incorporated into Middle-brook 7H11 agar medium with oleic acid, albumin, dextrose, and catalase (OADC) growth supplement to get final concentrations of 50, 25, 12.5, 6.25, 3.13, 1.56, and 0.78 μg mL−1. Inoculum of M. tuberculosis H37Rv ATCC 27294 was prepared from fresh Middlebrook 7H11 agar slants with OADC (Difco) growth supplement adjusted to 1 mg/mL (wet weight) in Tween 80 (0.05%) saline diluted to 10−2 to give a concentration of ∼107 cfu per mL−1. Five microliters of this bacterial suspension was spotted onto 7H11 agar tubes containing different concentrations of the drug as discussed above. The tubes were incubated at 37 °C, and final readings (as MIC in μg mL−1) were determined after 28 days. The MIC is defined as the minimum concentration of compound required to give complete inhibition of bacterial growth. This method is similar to that recommended by the National Committee for Clinical Laboratory Standards for the determination of MIC in triplicate.In vitro cytotoxicity screening
Antitubercular active compounds with MIC ≤ 12.5 μg mL−1 were examined for toxicity in a HEK-293T cellline at the concentration of 50 μg mL−1. After 72 h of exposure, viability was assessed on the basis of cellular conversion of (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) into a formazan product using the Promega Cell Titer 96 non-radioactive cell proliferation assay.Acknowledgements
Computations have been made available thanks to the Calcul Quebec, and Compute Canada.Notes and references
- World Health Organization, Global tuberculosis report (in IRIS), World Health Organization, Geneva, 2012.
- H. Mc Shane, Int. J. STD AIDS, 2005, 16, 95 CrossRef PubMed.
- (a) M. D. Scott, Biochem. Pharmacol., 2006, 71, 1096 CrossRef PubMed; (b) D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6, 29 CrossRef CAS PubMed; (c) L. P. Ormerod, Thorax, 1999, 54, 42 CrossRef.
- (a) K. Arya, P. Tomarand and J. Singh, RSC Adv., 2014, 4, 3060 RSC; (b) C. E. P. Galvis and V. V. Kouznetsov, Org. Biomol. Chem., 2013, 11, 7372 RSC; (c) A. Dandia, A. K. Jainand and A. K. Laxkar, RSC Adv., 2013, 3, 8422 RSC; (d) N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 RSC; (e) N. Hara, S. Nakamura, M. Sano, R. Tamura, Y. Funahashi and N. Shibata, Chem.–Eur. J., 2012, 18, 9276 CrossRef CAS PubMed; (f) N. V. Hanhan, Y. C. Tang, N. T. Tran and A. K. Frank, Org. Lett., 2012, 14, 2218 CrossRef CAS PubMed; (g) K. Ohmatsu, M. Kiyokawa and T. Ooi, J. Am. Chem. Soc., 2011, 133, 1307 CrossRef CAS PubMed; (h) S. W. Duan, J. An, J. R. Chen and W. J. Xiao, Org. Lett., 2011, 13, 2290 CrossRef CAS PubMed; (i) B. Tan, N. Candeias and C. F. Barbas III, Nat. Chem., 2011, 3, 473 CAS; (j) K. Shen, X. Liu, G. Wang, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4684 CrossRef CAS PubMed; (k) G. S. Singhand and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef PubMed; (l) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed.
- K. Atul, M. R. Awatar, S. A. Kumar, S. A. Bahadur and T. A. Kumar, Indian Pat. Appl., 2011, 48, 20110114 Search PubMed.
- A. Kumar, G. Gupta, S. Srivastava, A. K. Bishnoi, R. Saxena, R. Kant, R. S. Khanna, P. R. Maulikand and A. Dwivedi, RSC Adv., 2013, 3, 4731 RSC.
- G. Wu, L. Ouyang, J. Liu, S. Zeng, W. Huang, B. Han, F. Wu, G. He and M. Xiang, Mol. Diversity, 2013, 17, 271 CrossRef CAS PubMed.
- S. Sivakumar, R. R. Kumar, M. A. Ali and T. S. Choon, Eur. J. Med. Chem., 2013, 65, 240 CrossRef CAS PubMed.
- (a) A. I. Almansour, S. Ali, M. A. Ali, R. Ismail, T. S. Choon, V. Sellappan, K. k. Elumalai and S. Pandian, Bioorg. Med. Chem. Lett., 2012, 22, 7418 CrossRef CAS PubMed; (b) S. M. Rajesh, S. Perumal, J. C. Menendez, P. Yogeeswari and D. Sriram, Med. Chem. Commun., 2011, 2, 626 RSC; (c) P. Prasanna, K. Balamurugan, S. Perumal, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2010, 45, 5653 CrossRef CAS PubMed; (d) S. V. Karthikeyan, B. D. Bala, V. P. A. Raja, S. Perumal, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2010, 20, 350 CrossRef CAS PubMed; (e) S. U. Maheswari, K. Balamurugan, S. Perumal, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2010, 20, 7278 CrossRef CAS PubMed; (f) R. Kumar, S. Perumal, S. C. Manju, P. Bhatt, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2009, 16, 3461 CrossRef PubMed.
- (a) M. Isaka, N. Rugseree, P. Maithip, P. Kongsaeree, S. Prabpai and Y. Thebtaranonth, Tetrahedron, 2005, 61, 5577 CrossRef CAS PubMed; (b) T. Matviiuk, G. Morid, C. Lherbeta, F. Rodriguez, M. R. Pasca, M. Gorichko, B. Guidettia, Z. Voitenko and M. Baltasa, Eur. J. Med. Chem., 2014, 71, 46 CrossRef CAS PubMed.
- (a) K. Revathy and A. Lalitha, RSC Adv., 2014, 4, 279 RSC; (b) A. A. Shvets, Y. V. Nelyubina, K. A. Lyssenko and S. V. Kurbatov, Russ. Chem. Bull., Int. Ed., 2012, 61, 1659 CrossRef CAS.
- L. Yan, W. Yang, L. Li, Y. Shen and Z. Jiang, Chin. J. Chem., 2011, 29, 1906 CrossRef CAS.
- (a) K. Arora, S. Verma, D. Jose, P. Pardasani and R. T. Pardasani, Phosphorus, Sulfur Silicon, 2008, 183, 1168 CrossRef CAS; (b) R. T. Pardasani, P. Pardasani, A. Jain and K. Arora, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2006, 45, 1204 Search PubMed.
- K. N. Houk, Acc. Chem. Res., 1975, 8, 361 CrossRef CAS.
- (a) K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS; (b) K. Fukui, Acc. Chem. Res., 1971, 4, 57 CrossRef CAS; (c) K. Fukui, in Molecular Orbitals in Chemistry, Physics, and Biology, Academic, New York, NY, 1964, p. 525 Search PubMed.
- (a) M. Cossi, V. Barone, R. Cammi and J. Tomasi, J. Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (b) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS PubMed; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61 CrossRef CAS.
- (a) C. D. Valentin, M. Freccero, R. Gandolfi and A. Rastelli, J. Org. Chem., 2000, 65, 6112–6120 CrossRef PubMed; (b) K. N. Houk, J. Gonzalez and Y. Li, Acc. Chem. Res., 1995, 28, 81 CrossRef CAS.
- K. Chandraprakash, M. Sankaran, C. Uvarani, R. Shankar, A. Ata, F. Dallemer and P. S. Mohan, Tetrahedron Lett., 2013, 54, 3896 CrossRef CAS PubMed; P. Yuvaraj and B. S. R. Reddy, Tetrahedron Lett., 2013, 54, 821 CrossRef PubMed.
- M. Mujahid, R. G. Gonnade, P. Yogeeswari, D. Sriram and M. Muthukrishnan, Bioorg. Med. Chem. Lett., 2013, 23, 1416 CrossRef CAS PubMed.
- M. J Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1; Gaussian, Wallingford CT Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785 CrossRef; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- (a) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS PubMed; (b) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed; (c) P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209 CrossRef CAS; (d) M. S. Gordon, Chem. Phys. Lett., 1980, 76, 163 CrossRef CAS; (e) M. M. Franci, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. De Frees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654 CrossRef PubMed; (f) V. A. Rassolow, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 990193. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra11940a |
| This journal is © The Royal Society of Chemistry 2014 |