
A corannulene-based donor–acceptor polymer for organic field-effect transistors†
Abstract
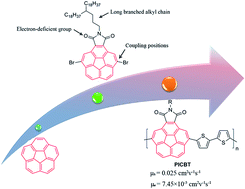
For the first time, the corannulene unit was incorporated directly into the backbone of conjugated polymers. A new donor–acceptor (D–A) copolymer PICBT using imide-fused corannulene as acceptor was synthesized and its performance in organic field-effect transistors (OFETs) was tested. PICBT exhibited ambipolar transporting property with a hole mobility of 0.025 cm2 V−1 s−1 and electron mobility of 7.45 × 10−5 cm2 V−1 s−1 when the substrates were treated with octyltrimethoxysilane (OTS). If the substrates were not modified with OTS, PICBT showed lower device performances with a hole mobility of 4.62 × 10−3 cm2 V−1 s−1 and electron mobility of 1.54 × 10−4 cm2 V−1 s−1. The device performances are competitive among the amorphous materials. This work paved the way for incorporating the corannulene unit into conjugated materials.
 Please wait while we load your content...
Please wait while we load your content...

