
A signal-on electrochemiluminescence aptasensor based on the quenching effect of manganese dioxide for sensitive detection of carcinoembryonic antigen†
Abstract
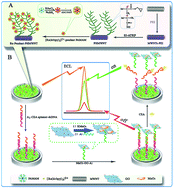
We developed a signal-on electrochemiluminescence (ECL) aptasensor by using SI-ATRP to facilitate high-density immobilization of luminophores and manganese dioxide–graphene (MnO2–GO) composite to indirect deactivate the excited state of Ru(dcbpy)32+ for ultrasensitive detection of carcinoembryonic antigen (CEA). In this approach, manganese dioxide–graphene (MnO2–GO) composite served as an efficient quencher for indirect deactivating the excited state of Ru(dcbpy)32+. Surface initiated atom transfer radical polymerization (SI-ATRP) was applied to functionalize multiwalled carbon nanotubes (MWNTs) with glycidyl methacrylate (GMA) as the functional monomer. A nanocomposite material of polyamidoamine (PAMAM) dendrimer encapsulated AuNPs was used as the carrier to combine Ru(dcbpy)32+ and poly-GMA together for the synthesis of the ECL matrices. The prepared matrices were applied to bind amino-modified auxiliary probe I (A1), which was partially complementary with the CEA aptamer. Meanwhile, the MnO2–GO composite was modified with another amino-modified CEA aptamer-partial-complementary auxiliary probe II (A2). Through the hybridization of CEA aptamer with A1 and A2, the quencher MnO2–GO composite was linked with the ECL matrices, by which a low ECL signal was detected (off-state). However, in the presence of CEA, the sandwich-like structure was destroyed because CEA would bind to its aptamer in lieu of the auxiliary probes, which resulted in a recovery of ECL signal (on-state). The proposed ECL aptasensor showed high sensitivity with a detection limit of 25.3 fg mL−1 and a wide linear range of 0.1 pg mL−1 to 20 ng mL−1. Consequently, with the excellent sensitivity, stability and satisfying precision, the as-proposed strategy constitutes a promising detection technique for clinical diagnosis.
 Please wait while we load your content...
Please wait while we load your content...

