
An efficient approach to the ammoxidation of alcohols to nitriles and the aerobic oxidation of alcohols to aldehydes in water using Cu(ii)/pypzacac complexes as catalysts†
Abstract
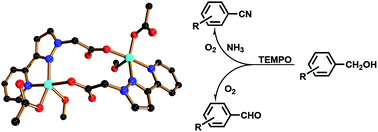
Reactions of 2-(3-(pyridin-2-yl)-1H-pyrazol-1-yl)acetic acid (pypzacacH) ligand with Cu(OAc)2, Cu(NO3)2, CuSO4, Cu(ClO4)2 or CuCl2 produced four dinuclear Cu(II) complexes [{(MeOH)Cu(OAc)}(μ-κ2:κ1-pypzacac)]2·0.5H2O (1·0.5H2O), [{Cu(pypzacac)}(μ-κ2:κ1-pypzacac)2{Cu(H2O)2}](NO3)·2(MeOH)0.5·6H2O (2·2(MeOH)0.5·6H2O), [{(MeOH)Cu(mpypzacac)}(μ-SO4)]2·2MeOH (3·2MeOH; mpypzacac = methyl 2-(3-(pyridin-2-yl)-1H-pyrazol-1-yl)acetate), [{Cu(mpypzacac)2}(μ-κ2:κ1-pypzacac){Cu(mpypzacac)}](ClO4)3·MeOH (4·MeOH) and one polymeric Cu(II) complex [(CuCl)(μ-κ3:κ1-pypzacac)]n (5), respectively. The mpypzacac ligand in 3 and 4 was in situ generated via the Cu2+-catalyzed dehydrative esterification of acetic acid of the pypzacacH ligand. Complexes 1–5 are characterized by elemental analysis, IR and single-crystal X-ray diffraction. Complex 1 contains two {(MeOH)Cu(OAc)} fragments that are interconnected by two μ-κ2:κ1-pypzacac− ligands, forming a dimeric structure. In 2, {Cu(pypzacac)} and {Cu(H2O)2} units are bridged by a pair of μ-κ2:κ1-pypzacac− ligands. In 3, two {Cu(mpypzacac)} fragments are linked by two μ-κ1:κ1-SO42− ions to form a dinuclear structure. Complex 4 also adopts a dimeric structure in which {Cu(mpypzacac)2} and {Cu(mpypzacac)} units are interconnected by one μ-κ3:κ1-pypzacac− ligand. Complex 5 contains a 1D chain in which (CuCl) fragments are interlinked by μ-κ3:κ1-pypzacac− ligands. Complexes 1–5 exhibited excellent catalytic performance in the ammoxidation of alcohol to nitrile and the aerobic oxidation of alcohol to aldehyde in water. The catalytic aqueous solution was easily separated and could be reused for several cycles without any obvious decay of catalytic efficiency.
 Please wait while we load your content...
Please wait while we load your content...

