
Noncovalently grafting sulfonic acid onto graphene oxide for improved hole transport in polymer solar cells†
Abstract
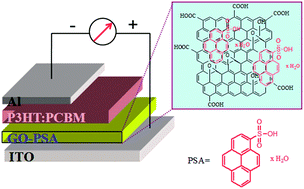
Sulfonic acid was successfully grafted onto graphene oxide (GO) via a facile noncovalent functionalization approach using pyrene as the anchoring bridge, affording a novel water-processable sulfonic acid functionalized GO, which shows improved hole transport in polymer solar cells (PSCs) compared to that of pristine GO. The successful grafting of 1-pyrenesulfonic acid (PSA) onto the carbon basal plane of GO is confirmed by FT-IR, UV-vis and Raman spectroscopic studies. An AFM study on the film morphology of GO–PSA reveals that the PSA moiety attaches onto both sides of the single layered graphene sheets, thus prohibiting the exfoliated single-layer graphene sheets from re-stacking. Finally, GO–PSA was applied as an effective hole transport layer (HTL) in poly(3-hexylthiophene-2,5-diyl):[6,6]-phenyl-C61-butyric acid methyl ester (P3HT:PCBM) bulk heterojunction (BHJ) PSC devices. Under the optimized conditions, the ITO/GO–PSA/P3HT:PCBM/Al BHJ-PSC device has a power conversion efficiency (PCE) of 2.86%, which is enhanced by ca. 42.3% compared to that of the reference device based on pristine GO HTL (2.01%). The PCE enhancement is primarily attributed to the increase of fill factor (FF) due to the improved hole transport of GO upon PSA grafting, which results from the improved conductivity of GO upon PSA grafting and the decrease of the contact resistance between P3HT and GO because of the enhanced surface doping of P3HT by the –OSO3H groups.
 Please wait while we load your content...
Please wait while we load your content...

