DOI:
10.1039/C4RA11653D
(Paper)
RSC Adv., 2014,
4, 65127-65136
Enhancing the sorption performance of surfactant-assisted CaO nanoparticles
Received
2nd October 2014
, Accepted 7th November 2014
First published on 7th November 2014
Abstract
Microsized calcium oxide prepared via precipitation and thermal decomposition of calcium carbonates has been widely used in industrial hydrogen production and biomass gasification processes to remove CO2 from the reactors. One of the most interesting perspectives in catalysis is the development of nano-sized, high performance, low cost catalysts. However, due to the high cost of nano-sized calcium oxide (CaO), it is critically important to develop new techniques that overcome this challenge. The main goal of this study was to prepare zwitterionic surfactant (BS-12) modified nano calcium oxide sorbents. Ctenocardia fornicata shell was used as the precursor for nano CaO, as it is cheap and easily available. The effect of BS-12 on the physico-chemical properties and the performance of the nano CaO sorbent for CO2 capture were investigated. Thermo gravimetric analysis (TGA), X-ray diffraction (XRD), Fourier transform-infrared (FT-IR), X-ray fluorescence (XRF), N2 adsorption–desorption (BET) measurements, transmission electron microscopy (TEM), zeta potential and temperature programmed desorption (TPD-CO2) studies were used for the characterization of the prepared nano-sized CaO particles. The results showed that BS-12 modified nano CaO exhibited the best performance for CO2 capture. The particle size and morphology of CaO varied from rod shape (45–33 nm) to cubic (13–23 nm) by changing the operating variables. A suggested mechanism for nanoparticle formation in the presence of BS-12 is also discussed.
Introduction
In combating climate change, CO2 capture and storage (CCS) using various CO2 absorbents represents the sole effective solution to reduce such a huge amount of CO2. The emissions of CO2 can be reduced by CO2 capture in industrial hydrogen production processes such as coal gasification, methane steam reforming, water gas shift reaction and biomass gasification.1 In addition to the environmental impact, CO2 removal from the reactor in the biomass gasification process improves the volatilisation and gasification steps, shifting the balance towards hydrogen production.2 CO2 is captured in the form of CaCO3. However, the carbonation and calcination reactions are not totally reversible but are related to the conditions, such as temperature and pressure,3 as follows:
Indeed, calcium oxide (CaO) readily obtained through the calcination of naturally occurring sea shells has been proposed as an alternative CO2 sorbent that could substantially reduce the costs of CO2 capture. Thus, the benefits associated with the use of nano calcium carbonate (CaCO3) as a CaO precursor can be expected to create a positive breakthrough.
There are very few reports on the preparation of CaO in the nano size range. Koper et al.4 and Tang et al.5 both used a sol–gel method to prepare nano CaO from calcium methoxide (Ca(OCH3)2) and calcium nitrate (Ca(NO3)2·4H2O) precursors, respectively. Recently, Amin Alavi and Morsali6 used a sonochemical method to synthesize CaO from calcium acetate and nanoparticle tetramethylammonium hydroxide precursors. More recently, Roy and Bhattacharya7 performed a microwave irradiation technique to synthesize nano CaO particles using Ca(NO3)2·4H2O and NaOH. However, the cost associated with the raw materials is one of the drawbacks of these processes. While other studies have investigated the effect of the type of surfactant on the crystallization and polymorphic transitions of emulsified particles,8–11 a study on the effect of zwitterionic surfactants on nanoparticle formation and their performance for CO2 capture has, to the best of our knowledge, not yet been conducted. Ctenocardia fornicata shells were used as a precursor of nano CaO, as they are cheap and easily available. A suggested mechanism for nanoparticle growth in the presence of surfactant is also discussed.
Experimental
Materials
Ctenocardia fornicata shell waste was collected from Port Dickson beach, Negeri Sembilan, Malaysia. Dodecyl dimethyl betaine (BS-12) was purchased from Sigma-Aldrich (Steinheim, Germany). The deionized water used was HPLC-grade of resistance >18 MΩ obtained from a Milli-Q-Water System (Organex). All the chemical reagents used in these experiments were of analytical grade.
Sorbent preparation
The nano-sorbent CaO was prepared from Ctenocardia fornicata shells. The shells were washed with deionized water to remove the dirt and dried at 110 °C in the oven for 2 h. Then, the shells were finely grounded using a blender (Blendor, HCB 550, USA). The powders were sieved using a stainless laboratory test sieve with an aperture size of 250 μm (Endecott Ltd., London, England) to obtain micron-sized powders. In a typical procedure, 10 g of micron-sized Ctenocardia fornicata shell powder was stirred with 100 mL deionized water in a conical flask to from a colloidal solution. The pH of the solution was adjusted to 6.4–6.5 to facilitate the decomposition of CaCO3.12 Subsequently, BS-12 (4%) was added into the colloidal solution and the pH was adjusted to 13.5 with 10 N NaOH. The resulting colloidal solutions were then stirred (1000 rpm) for 30 (S1), 60 (S2), 90 (S3) and 120 (S4) min at room temperature. The obtained samples were separated using double ring filter paper and dried at 100 °C in the oven for 24 h. Finally, the dried samples were calcined at 850 °C for 4 h. The calcined samples were labelled as C1, C2, C3 and C4, corresponding to stirring time intervals of 30, 60, 90 and 120 min, respectively.
Sorbent characterization
Thermogravimetric. Thermogravimetric analysis (TGA) of the waste cockle (Ctenocardia fornicata) shell powder was employed on a Mettler Toledo thermogravimetric analyser. The analysis was performed in a nitrogen atmosphere over a temperature range of 35–1000 °C at a heating rate of 10 °C min−1.
Structure and crystallography. X-ray powder diffraction (XRD) analysis of the sample was carried out using a Powder X-ray diffractometer (Shidmazu Corporation, Japan) model XRD 6000 in reflection mode with Cu Kα radiation. The Cu Kα radiation was generated by a Philips glass diffraction X-ray tube (broad focus 2.7 kW type).
Surface functional group. The surface functional group of the sample was ascertained using attenuated total reflection-Fourier transform-infrared (FT-IR) on a Perkin Elmer (PC) Spectrum 100 FTIR spectrometer. The FT-IR spectra were obtained over the region 400–4000 cm−1 with spectral resolution of 4 cm−1.
Elemental analysis. The chemical composition of the samples was determined by energy dispersive X-ray fluorescence (XRF) spectrometry using a Bruker AXS. The metal concentration in the examined samples was determined by the amount of emitted X-ray radiation in relation to the values in the calibration curves.
Specific surface area. The BET specific surface area of the sample was measured by the Brunauer–Emmett–Teller (BET) method using nitrogen adsorption at −196 °C. The analysis was conducted using a Surfur, Thermo Scientific instrument.
Morphology. The morphological observation of the sample was made by Transmission Electron Microscopy (TEM, Hitachi, H7100). TEM specimens were prepared by depositing a few drops of sample dispersed in ethanol on a carbon coated copper grid. Particle size of the sample was obtained by using TEM with an accelerating voltage of 200 kV.
Surface charge analysis. Zeta potential measurement was performed by a 3000 HSA analyzer (Malvern). The suspension was made by the self-dispersed powder in methanol. Light scattering was monitored at a 90° scattering angle and a temperature of 25 °C in a clear disposable cell.
Sorbent sorption capability. The absorption test of the sorbents was carried out by CO2 pulse chemisorption and temperature programmed desorption (CO2-TPD) analysis. Both experiments were performed using a Temperature Programmed chemisorption–desorption (Thermo Finnigan TPDRO 1000) apparatus equipped with a thermal conductivity detector (TCD). Prior to the pulse chemisorption experiment, 200 mg of synthesized sorbent was pretreated at 750 °C for 30 min under a helium stream (10 °C min−1, 20 mL min−1) in order to desorb CO2 and moisture molecules that come from the atmospheric air, and then the temperature was maintained at 750 °C. Subsequently, the measurement of CO2 absorption was recorded continually during the entire process at 750 °C under pure CO2 gas (20 mL min−1) for 1 h. The absorption of CO2 on the sorbent was evaluated from the peak areas detected by TCD.For CO2-TPD measurements, 200 mg of sorbent was treated at its calcination temperature (850 °C) under a helium flow rate of 30 mL min−1 for 1 h, and saturated with a pure CO2 flow (30 mL min−1) after cooling to 60 °C. Then, it was flushed with helium (30 mL min−1) to remove weakly physisorbed CO2. Consequently, the sorbent was proceed at temperature between 50-900 °C under helium (10 °C min−1, 30 mL min−1) as carrier gas and hold for 10 min. Afterward, the CO2 desorption signal was recorded by TPD with the linear temperature increased up to 900 °C at rate of 10 °C min−1. The area of the desorption profiles was obtained: the total desorption of CO2 at 900 °C is then equivalent to the total CO2 that could be absorbed in the sorbent.
Results and discussion
Previous studies have reported that the addition of surfactant can inhibit crystal growth due to the formation of an energy barrier by the surfactant at the solid–liquid interface.13 It should be pointed out that any of the atoms in the nanoparticles can act as the absorber atom.14 A strong absorption of surfactant at the nanoparticle growth site would reduce the growth rate of nanoparticles, as reported by Mehta et al.,15 although Sajanlal et al.16 concluded from their work that full coverage with surfactant would hinder the diffusion of growth species from the surrounding solution to the surface of the growing particles. Hence, the synthesis of CaO nanoparticles from Ctenocardia fornicata shells using a surfactant-assisted precipitation approach is proposed in this study. In their discussion of the decomposition properties of calcium carbonate, Tai et al.17 have emphasized the influence of pH on the decomposition. A similar conclusion was reached by Han et al.,12 who reported that the decomposition of CaCO3 could be facilitated at a pH below 6.5. It should be noted that the zwitterionic surfactant (BS-12) used in this study possesses polar head groups which, on ionisation, impart both positive and negative charges at the isoelectric point. As the pH shifts away from the isoelectric point to 13.5, the molecule of surfactant behaves as an anionic surfactant.18 Thus, the precipitation of calcium carbonate could take place at this pH due to the electrostatic repulsion interactions between the anionic surfactant and the anionic carbonate ions. Finally, CaO sorbent was produced via thermal decomposition of precipitated calcium carbonate. As indicated earlier, the objective of the present work was to compare the sorption performance of surfactant-assisted CaO nanoparticles, and this, in turn, required the preparation of well-characterized CaO nanoparticles.
Thermal decomposition of sorbents
Ctenocardia fornicata shells were analyzed by TGA to determine the decomposition temperature of the shells. Fig. 1 shows the TGA curves for the Ctenocardia fornicata shell and the treated shells. The thermogram (Fig. 1a) of the shell shows two weight loss steps with a total weight loss of 56.7%. A single major weight loss peak was observed from the TGA curve at a temperature range of 600–800 °C (47%), which could be attributed to the decomposition of CaCO3 into CaO. This is in agreement with the report by Sivakumar et al.19 The weight of the sample remained constant at above 850 °C. Therefore, complete decomposition occurred at temperatures above 850 °C, required to transform CaCO3 into CaO as follows:
| CaCO3(s) → CaO(s) + CO2(g) |
 |
| | Fig. 1 TGA analyses of the powdered Ctenocardia fornicata shell (a) and treated shells: (b) S1 (c) S2, (d) S3 and (e) S4. | |
Fig. 1b–e shows that the initial decomposition temperature of the treated shells decreased gradually, which suggests that the grain size of the treated shell was reduced by the treatment with the surfactant. Archer Jr et al.20 studied the thermal decomposition kinetics of calcium carbonate and reported that the decomposition of CaCO3 at low temperatures is characteristic of small-sized particles. Further, it has been argued by Zuiderduin et al.21 that large particles display a higher decomposition temperature than small particles.
Structure of sorbents
The XRD pattern of Ctenocardia fornicata shell shown in Fig. 2a suggest the presence of the CaCO3 phase, without any additional diffraction peaks, at 2θ values of 26.3°, 27.3°, 31.2°, 33.1°, 36.2°, 37.9°, 38.7°, 41.3°, 43.0°, 45.9°, 48.4°, 50.2°, 52.5°, 53.1°, 66.2° and 69.0° (JCPDS file: 01-071-2396). The diffraction pattern of treated shell S-4 is similar to Fig. 2a. The XRD profiles of the sorbents (Fig. 2c–f) correspond to the characteristic CaO peaks at 2θ values of 32.1°, 37.3°, 53.8°, 64.1°, 67.3° and 79.6° (JCPDS file: 00-037-1497). No peaks from any other phases of CaO were observed. The results indicate that the thermal decomposition of CaCO3 in the powder shell is converted into CaO, which agrees with the results from the literature.19,22 Although the peak positions in the XRD pattern in Fig. 2c–f are similar, the XRD peaks of these compounds gradually appear broader suggesting a decrease of the crystallite size.23
 |
| | Fig. 2 X-ray diffraction pattern of (a) Ctenocardia fornicata shell, (b) treated shell S4 and sorbents: (c) C1, (d) C2, (e) C3 and (f) C4. | |
Surface functional groups of the sorbents
The FT-IR spectra of Ctenocardia fornicata shell, the treated shell and the sorbents (C1–C4) are presented in Fig. 3 and Table 1. The IR spectrum of the shell (Fig. 3a) shows bands at 1455, 857 and 712 cm−1, characteristic of the C–O stretching and bending modes of calcium carbonate.24 In addition, the weak band at 1794 cm−1 is assigned to an overtone or combination of some vibrational modes of divalent metal ions and the carbonate group bond. Although the absorption peaks of the surfactant treated shell (Fig. 3b) are similar, the existence of alkyl group bands at 2983 and 2533 cm−1 are due to the hydrophobic tail of the surfactant, formed during the treatment. Fig. 3c–f shows the spectrum of the calcined treated shell. The results shows that the spectra of all CaO sorbents (Fig. 3c–f) appear at the same wave number, indicating that they have a very similar chemical structure. The existence of a band at 3640 cm−1 is due to the OH groups in the Ca(OH)2 formed during the adsorption of water by CaO. The wide and strong band at around 500 cm−1 corresponds to the Ca–O bond.
 |
| | Fig. 3 FT-IR spectra of (a) Ctenocardia fornicata shell, (b) treated shell S4, and sorbents (c) C1, (d) C2 (e) C3 and (f) C4. | |
Table 1 Surface functional groups of Ctenocardia fornicata shell, treated shell and sorbents
| |
Peak positiona (cm−1) |
| Assignment |
3640 |
2983 |
2533 |
1794 |
1455 |
1182 |
857 |
712 |
500 |
| OH |
|
|
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
CO32− |
C–N |
CO32− |
CO32− |
Others |
| s = strong, m = medium and w = weak intensities; NA = not found. |
| Sample |
|
|
|
|
|
|
|
|
|
| Ctenocardia fornicate |
NA |
NA |
NA |
w |
s |
m-s |
s |
m-s |
s |
| S4 |
NA |
w |
w |
w |
s |
m-s |
s |
m-s |
s |
| C1 |
w |
NA |
NA |
NA |
W |
NA |
w |
NA |
s |
| C2 |
w |
NA |
NA |
NA |
W |
NA |
w |
NA |
s |
| C3 |
w |
NA |
NA |
NA |
W |
NA |
w |
NA |
s |
| C4 |
w |
NA |
NA |
NA |
W |
NA |
w |
NA |
s |
Composition of Ctenocardia fornicata
The XRF analysis results (Table 2) confirmed that the composition of the Ctenocardia fornicata shell mainly consists of Ca (99.3%). The high amount of Ca is associated with the presence of CaCO3, which is the main component of the shell, as confirmed by the XRD and FT-IR results. The shell contains small amounts of S, K, Sr, Cu and Fe. Thus, the shell can be considered, from a chemical viewpoint, as a relatively pure natural carbonate-based material.
Table 2 Chemical composition of Ctenocardia fornicata shell
| Metal composition |
Percentage (%) |
| Ca |
99.3 |
| S |
0.345 |
| K |
0.238 |
| Sr |
0.082 |
| Cu |
0.048 |
| Fe |
0.027 |
Surface charge of calcium carbonate
Fig. 4 and Table 3 show the zeta potentials of the Ctenocardia fornicata shell and the treated shell S4. It can be observed that the zeta potential of the BS-12 treated sample (S4) at pH 6.4 is −20.13 mV (Fig. 4a), whereas, this value decreased to −4.13 mV at pH 13.5 (Fig. 4b). Thus, these results reveal that BS-12 neutralizes the surface charge of CaCO3 at pH 13.5 and the zeta potential approaches to zero.
 |
| | Fig. 4 Zeta potential of the treated Ctenocardia fornicata shell (S4) at (a) pH 6.4 and (b) pH 13.5. | |
Table 3 Zeta potential of the Ctenocardia fornicata shell and the treated shell
| pH |
Zeta potential (mV) |
| Ctenocardia fornicate shell |
Treated sample (S4) |
| 6.4 |
−18.61 |
−20.13 |
| 13.5 |
−11.13 |
−4.13 |
Size and morphology of sorbents
Fig. 5 shows representative TEM images of the sorbents. Rod-shaped CaO nanoparticles with a diameter of about 33–45 nm are predominant after 30 min of retention time (Fig. 5a), while regular and spherical ones with a particle size of 30–37 nm are prevailing over a time course of 60 min (Fig. 5b). When the reaction time is up to 90 min, the size of the spherical particles is reduced to 24–30 nm (Fig. 5c). Smaller nanoparticles can be obtained by further increasing the period of time up to 120 minutes, (Fig. 5d) for which the average diameter of the CaO nanorods is about 13–23 nm. The produced particles have a narrow size distribution and limited amount of agglomerates. The results reveal that the surfactant treatment for different time periods affect the shape and size of the CaO nanoparticles. It has been reported by El-Sheikh et al.25 that surfactant molecules attach to the solid particle and form a layer between the particle and the surrounding fluid molecules. Such layers impart a repulsive force between them and, in turn, prevent particle aggregation. A conclusion has been reached by Tanvir and Qiao26 that the surfactant increases the electrostatic force of the solid–liquid interface and thus reduces the aggregation of particles. A similar observation has been made by Helgason et al.27 who investigated the impact of surfactants on the rate of crystal growth of solid lipid nanoparticles. Further, it has been shown by Idrees28 that the strong adsorption of surfactant would occupy the nanoparticle growth sites, thus reducing the growth of particles. Thus, the formation of nanoparticles observed in this study seems to be indicative of a nucleation sensitive phenomenon which is presumably an inherent feature of the zwitterionic surfactant (BS-12). Therefore, the presence of a zwitterionic surfactant, besides a large CaCO3 nucleation rate in the initial period of the carbonation reaction, controls the rate of crystal growth and therefore, monodisperse CaCO3 nanoparticles are successfully prepared.
 |
| | Fig. 5 Transmission electron microscopy images of sorbents: (a) C1, (b) C2, (c) C3; (d) C4. | |
Proposed mechanism
The main ingredient in the Ctenocardia fornicata shell is CaCO3, which was confirmed by XRD, FT-IR, XRF and TGA analyses. Calcium carbonate aqueous solution pH value is 8.0–8.6. The pH of the solution was adjusted below 6.5 to facilitate the decomposition of CaCO3 (eqn (1)–(4)).12 Han et al.12 reported that the reaction in eqn (3) is slower than the reaction in eqn (2). Hence, CaCO3 is not precipitated at pH < 6.5 due to the absence of sufficient CO32− ions in solution.12 It is well known that dodecyl dimethyl betaine (BS-12) is an amphoteric surfactant, containing both cationic and anionic groups (eqn (5)). Addition of BS-12 to a colloidal suspension of CaCO3 at pH 13.5 allows BS-12 to dissociate dodecyldimethylaminoacetate (eqn (6)). The dissociation of CaCO3 at pH 13.5, as reported by Han et al.,12 is shown in eqn (7) and (8). The reaction in eqn (8) is faster than the reaction in eqn (7) at pH 13.5, which leads to an even higher concentration of CO32− than of HCO3− in solution.12 Therefore, precipitation of CaCO3 can be achieved at pH over 13.5 according to the reactions in eqn (9) and (10). This may be due to the electrostatic repulsion between the negatively charged alkyl chain and the CO32− ions, which makes their adsorption onto the face of CaCO3 difficult. Finally, calcium oxide is produced via thermal decomposition of calcium carbonate (eqn (11)). The suggested mechanism has been confirmed by TGA, XRD, FT-IR, zeta potential and TEM analyses, as shown in Fig. 1–5.| | |
CaCO3(s) + 2H2O → Ca(OH)2(aq) + H2CO3(aq)
| (1) |
| | |
H2CO3(aq) ⇌ HCO3(aq)− + H(aq)+
| (2) |
| | |
HCO3(aq)− ⇌ CO3(aq)2− + H(aq)+
| (3) |
| | |
Ca(OH)2(aq) ⇌ Ca(aq)2+ + 2OH−
| (4) |
| |
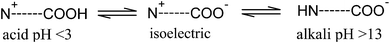 | (5) |
| | |
[CH3(CH2)11]N+(CH3)2CH2COOH + H2O → CH3(CH2)11NH(CH3)2CH2COO(aq)− + OH− + H+
| (6) |
| | |
CO2(aq) + OH(aq)− ⇌ HCO3(aq)−
| (7) |
| | |
HCO3(aq)− + OH(aq)− ⇌ H2O(l) + CO3(aq)2−
| (8) |
| | |
2[CH3(CH2)11]NH(CH3)2CH2COO(aq)− + Ca2+ → ([CH3(CH2)11]NH(CH3)2CH2COO−)2Ca(aq)2+
| (9) |
| | |
([CH3(CH2)11]NH(CH3)2CH2COO−)2Ca(aq)2+ + CO3(aq)2− → 2([CH3(CH2)11]NH(CH3)2CH2COO−)(aq) + CaCO3(s)
| (10) |
| |
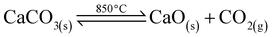 | (11) |
BET specific surface area and porosity of sorbents
Fig. 6 presents the N2 physisorption (adsorption–desorption) isotherms of the sorbents. According to the International Union of Pure and Applied Chemistry (IUPAC) classification of adsorption isotherms, type II describes the presence of mesopores (2–50 nm pore diameter) and micropores exhibit a hysteresis loop and a variation point at low pressure (P/P0) in a material.29 The isotherms have a sharp slope at relative pressures from 0 to 0.1, which can be attributed to the presence of micropores, and a small slope at relative pressures from 0.4 to 0.8, which indicates a broad pore-size distribution in the mesopore range (Fig. 6a–f). However, the surface area of each sample was quite different (Table 4).
 |
| | Fig. 6 N2 adsorption–desorption isotherms of (a) commercial CaO and sorbents (a) C1, (b) C2, (c) C3 and (d) C4. | |
Table 4 BET surface area analysis of commercial CaO and sorbents
| Composition |
BET surface area (m2 g−1) |
BJH desorption average pore diameter (nm) |
| Commercial CaO |
4.7 |
10.62 |
| C1 |
8.4 |
8.05 |
| C2 |
10.8 |
6.23 |
| C3 |
13.5 |
6.50 |
| C4 |
23.7 |
2.26 |
The pore size distribution curves, as shown in Fig. 7a–f, demonstrated a uni-modal pore size distribution for all sorbents. Commercial CaO (Fig. 5a) mainly exhibits pores of diameter larger than 10 nm while the C1, C2, C3 and C4 sorbents (Fig. 7b–f) present a distribution shifted towards smaller pore sizes (>2 nm). The BET surface areas of commercial CaO, C1, C2, C3 and C4 are 4.7, 8.4, 10.8, 13.5 and 23.7 m2 g−1, respectively, corresponding to average value pore sizes of 10.62, 8.05, 6.23, 6.50 and 2.26 nm, respectively. The results revealed that the surface area of the sorbent increases with increasing the length of stirring time. The C4 sorbent shows the highest specific surface area (24 m2 g−1), which is mainly a contribution of mesopores and micropore surfaces. The surface area data (SBET 5–23 m2 g−1) reported in Table 4 can be favourably compared to nano CaO produced from different shells.30,31
 |
| | Fig. 7 Pore size distribution curves of (a) commercial CaO and sorbents (b) C1, (c) C2, (d) C3 and (e) C4. | |
Performance of sorbents
CO2-TPD measurements were carried out to determine the absorption amount of CO2 in the treated and commercially available CaO sorbents (Fig. 8). The results of the CO2 absorption test are presented in Table 5. It is obvious that the treated samples present significant differences in CO2 sorption performance compared to commercially available CaO sorbents. These differences could be partially attributed to the higher BET surface areas of the treated sorbents compared to commercial sorbents, maximizing the number of available sorption sites for CO2 absorption. Sun et al.32 suggested that the rapid absorption of CO2 at the beginning of the process is due to external surface of the absorbent, and is followed by a slower internal diffusion process. According to the model developed by Bouquet et al.,33 the first step of carbonation finishes when all the grains are filled and the carbonate layer reaches a critical thickness; then, in the second step, CO2 must diffuse at the level of the whole grain. Further, it was reported by Lu and Smirniotis34 that the diffusion of CO2 could be accelerated by decreasing the particle size. Thus, nano CaO increases the exposed surface area for the surface reaction of CO2 and decreases the thickness of the CaO layer for CO2 diffusion. To have an insight into the CO2 absorption of the sorbents, a desorption test was performed using the same batch of sorbents (Fig. 9) and the results are shown in Table 5. The results show that the amount of desorbed CO2 is very close to the amount absorbed. Thus, this result confirms the connection between the physical properties of the sorbent and their absorption–desorption capacities. However, the sorption performance of CaO could be affected by the lattice structure of the sorbent, as reported by Ying et al.35 Thus, issues related to the lattice structure need to be addressed in the future.
 |
| | Fig. 8 CO2 absorption performance of (a) commercial CaO and sorbents (b) C1, (C) C2, (d) C3 and (e) C4. | |
Table 5 The amount of CO2 absorption and desorption of the sorbents
| Sample |
Sorption (mol g−1) |
| Absorptiona |
Desorptionb |
| Determined by chemisorption experiment. Determined by CO2-TPD analysis. |
| Commercial CaO |
256.17 |
245.33 |
| C1 |
683.10 |
654.56 |
| C2 |
783.93 |
703.77 |
| C3 |
854.47 |
813.38 |
| C4 |
1134.89 |
982.24 |
 |
| | Fig. 9 CO2 desorption performance of (a) commercial CaO and sorbents (b) C1, (C) C2, (d) C3 and (e) C4. | |
Conclusions
Nano calcium oxide sorbents having a diverse range of sizes and morphologies were synthesized in this work. The results suggest that the zwitterionic surfactant coverage at the interface of the particles controls the rate of crystal growth and favours the formation of nano calcium oxide. The produced calcium oxide particle morphology and size are significantly changed from rod shape (45–33 nm) to cubic (13–23 nm) depending on the operating variables. The synthesized nano sorbents were subjected to chemisorption–desorption experiments in the presence of CO2. The sorbents with higher surface area demonstrated a larger capacity for CO2 chemisorption–desorption. Among all the sorbents, C4 had the largest capacity for capturing CO2 due to its smaller CaO particle size, which provides a higher exposed surface area for the reaction with CO2. However, research related to the lattice structure needs to be addressed in the future, as it may affect the sorption performance of calcium oxide.
References
- A. A. Olajire, Energy, 2010, 35(6), 2610 CrossRef CAS PubMed.
- N. H. Florin and A. T. Harris, Chem. Eng. Sci., 2008, 63(2), 287 CrossRef CAS PubMed.
- F. Garcıa-Labiano, A. Abad, L. F. De Diego, P. Gayan and J. Adanez, Chem. Eng. Sci., 2002, 57(13), 2381 CrossRef.
- O. B. Koper, I. Lagadic, A. Volodin and K. J. Klabunde, Chem. Mater., 1997, 9(11), 2468 CrossRef CAS.
- Z. X. Tang, D. Claveau, R. Corcuff, K. Belkacemi and J. Arul, Mater. Lett., 2008, 62(14), 2096 CrossRef PubMed.
- M. Amin Alavi and A. Morsali, J. Exp. Nanosci., 2010, 5(2), 93–105 CrossRef CAS.
- A. Roy and J. Bhattacharya, Int. J. Nanosci., 2011, 10(03), 413 CrossRef CAS.
- H. Bunjes, M. H. Koch and K. Westesen, J. Pharm. Sci., 2003, 92(7), 1509 CrossRef CAS PubMed.
- H. Bunjes, F. Steiniger and W. Richter, Langmuir, 2007, 23(7), 4005 CrossRef CAS PubMed.
- T. Awad and K. Sato, J. Am. Oil Chem. Soc., 2001, 78(8), 837 CrossRef CAS PubMed.
- T. Awad and K. Sato, Colloids Surf., B, 2002, 25(1), 45 CrossRef CAS.
- S. J. Han, M. Yoo, D. W. Kim and J. H. Wee, Energy Fuels, 2011, 25(8), 3825 CrossRef CAS.
- M. M. Fryd and T. G. Mason, Soft Matter, 2014, 10, 4662 RSC.
- M. K. Hedayati, F. Faupel and M. Elbahri, Materials, 2014, 7(2), 1221 CrossRef CAS PubMed.
- S. K. Mehta, S. Kumar, S. Chaudhary and K. K. Bhasin, Nanoscale Res. Lett., 2009, 4(10), 1197 CrossRef CAS PubMed.
- P. R. Sajanlal, T. S. Sreeprasad, A. K. Samal and T. Pradeep, Nano Rev., 2011, 2, 5883 Search PubMed.
- C. Y. Tai, W. R. Chen and S. M. Shih, AIChE J., 2006, 52(1), 292 CrossRef CAS.
- S. Paria and K. C. Khilar, Adv. Colloid Interface Sci., 2004, 110(3), 75 CrossRef CAS PubMed.
- P. Sivakumar, P. Sivakumar, K. Anbarasu, R. Mathiarasi and S. Renganathan, Int. J. Green Energy, 2014, 11(8), 886 CrossRef CAS.
- P. D. Archer Jr, D. W. Ming and B. Sutter, Planet. Sci., 2013, 2, 2 CrossRef.
- W. C. J. Zuiderduin, C. Westzaan, J. Huetink and R. J. Gaymans, Polymer, 2003, 44(1), 261 CrossRef CAS.
- W. Suryaputra, I. Winata, N. Indraswati and S. Ismadji, Renewable Energy, 2013, 50, 795 CrossRef CAS PubMed.
- Y. Park, W. J. Lee and H. G. Kim, J. Phys.: Condens. Matter, 1997, 9(43), 9445 CrossRef CAS.
- G. Gergely, F. Wéber, I. Lukács, A. L. Tóth, Z. E. Horváth, J. Mihály and C. Balázsi, Ceram. Int., 2010, 36(2), 803 CrossRef CAS PubMed.
- S. M. El-Sheikh, S. El-Sherbiny, A. Barhoum and Y. Deng, Colloids Surf., A, 2013, 422, 44 CrossRef CAS PubMed.
- S. Tanvir and L. Qiao, Nanoscale Res. Lett., 2012, 7(1), 1 CrossRef PubMed.
- T. Helgason, T. S. Awad, K. Kristbergsson, D. J. McClements and J. J. Weiss, J. Colloid Interface Sci., 2009, 334(1), 75 CrossRef CAS PubMed.
- M. Idrees, Iran. J. Chem. Chem. Eng., 2013, 32(3), 27 Search PubMed.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone and K. K. Unger, Pure Appl. Chem., 1994, 66(8), 1739 CrossRef CAS.
- G. Montes-Hernandez, A. Fernandez-Martinez, L. Charlet, D. Tisserand and F. Renard, J. Cryst. Growth, 2008, 310(11), 2946 CrossRef CAS PubMed.
- L. Zhao, Z. Qiu and S. M. Stagg-Williams, Fuel Process. Technol., 2013, 114, 154 CrossRef CAS PubMed.
- P. Sun, J. R. Grace, C. J. Lim and E. J. Anthony, Energy Fuels, 2007, 21(1), 163 CrossRef CAS.
- E. Bouquet, G. Leyssens, C. Schönnenbeck and P. Gilot, Chem. Eng. Sci., 2009, 64(9), 2136 CrossRef CAS PubMed.
- H. Lu and P. G. Smirniotis, Ind. Eng. Chem. Res., 2009, 48(11), 5454 CrossRef CAS.
- Z. Ying, H. Jian Ming, C. Quan Zhen, Q. Meia, L. yia, H. Xin and Z. Yong Fan, Chin. J. Struct. Chem., 2013, 32(11), 1715 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 











