
Two novel ambipolar donor–acceptor type electrochromic polymers with the realization of RGB (red-green-blue) display in one polymer†
Abstract
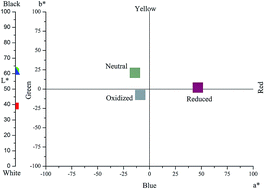
Two novel electrochromic monomers, 4,7-bis(4-methoxythiophen-2-yl)-[1,2,5]thiadiazolo[3,4-c]pyridine (MOTTP) and 4,7-bis(4-butoxythiophen-2-yl)-[1,2,5]thiadiazolo[3,4-c]pyridine (BOTTP), were synthesized and electropolymerized to give the corresponding polymers PMOTTP and PBOTTP, respectively. For the investigation of their electrochemical and electrochromic properties, the polymers were characterized using cyclic voltammetry (CV), UV-vis spectroscopy, step profiling, and scanning electron microscopy (SEM). The band gaps of the polymers were calculated based on spectroelectrochemical analysis, and were found to be 0.950 eV and 1.088 eV for PMOTTP and PBOTTP, respectively. Electrochromic investigations showed that PMOTTP and PBOTTP showed similar multichromic behaviors: saturated green color in the neutral state, highly transmissive blue in the oxidized state, and saturated red in the reduced state (red-green-blue, RGB). In addition, both polymers have excellent switching properties with more than 60% optical contrast in the NIR region and about a 0.5 s response time from neutral to oxide. Moreover, via electrochemical and spectral analyses both polymers were proven to be n-type dopable polymers. Hence, both polymers are promising materials to complete RGB electrochromic polymers for commercial applications.
 Please wait while we load your content...
Please wait while we load your content...

