
A facile strategy to enable nanoparticles for simultaneous phase transfer, folate receptor targeting, and cisplatin delivery†
Mochamad Zakki Fahmiab and
Jia-Yaw Chang*a
aDepartment of Chemical Engineering, National Taiwan University of Science and Technology, 43, Section 4, Keelung Road, Taipei 10607, Taiwan. E-mail: jychang@mail.ntust.edu.tw; Fax: +886-2-27376644; Tel: +886-2-27303636
bDepartment of Chemistry, Airlangga University, Jalan Mulyorejo, Surabaya 60115, Indonesia. E-mail: m.zakki.fahmi@fst.unair.ac.id; Fax: +62-31-5922427; Tel: +62-31-5922427
First published on 27th October 2014
Abstract
Diagnostic and therapeutic strategies related to cancer treatment are being continually developed for enhancing the effectiveness of the treatment. Active targeting nanoparticles (NPs) with therapeutic capability are a major requirement of cancer therapeutics. Herein, we present a general one-pot approach for simultaneously (1) achieving the phase transfer of various NPs from an organic solvent to an aqueous phase, and (2) grafting an active targeting ligand (folate) via electrostatic interactions as well as a therapeutic agent (cisplatin) via coordinative interactions of platinum atoms with carboxylic acid ligands on the surface of the NPs. This approach allows rapid, simple, and versatile bioconjugation without the need for additional crosslinking reagents. Taking AgInS2/ZnS quantum dots (AQDs) as an example, we show that NPs can be tailored to possess both targeting activity and therapeutic capability. In this study, AQDs exhibited a dose-dependent antiproliferative effect on HeLa cancer cells, indicating the possibility of using the obtained AQDs as highly effective dual-modality imaging probes for simultaneous cancer diagnosis and chemotherapy. In addition, the proposed one-pot approach can be extended to various other NPs (e.g., quantum dots, metal oxides, and metallic and magnetic materials) that can be successfully conjugated with folate via hydrophobic interactions and electrostatic immobilization during ultrasonication. Bioconjugation of the NPs was investigated by dynamic light scattering, ultraviolet-visible spectroscopy, and Fourier transform infrared spectroscopy before and after the phase transfer.
Introduction
Over the last decade, significant research has been conducted on nanoparticles (NPs), including quantum dots, as well as metal, metal oxide, or magnetic NPs, to investigate their possible applications in cancer diagnosis and therapeutics. These NPs are generally passivated using hydrophobic capping ligands because they are immiscible in the aqueous phase. For biological and medical applications, it is therefore essential to develop a general strategy for the phase transfer of NPs from an organic to aqueous medium. With regard to biologically motivated applications, it is also crucial to develop NPs that can carry therapeutic drugs and specifically deliver them into cancer cells for the diagnosis and/or treatment of different diseases.1 The effectiveness of cancer therapeutic nanocarriers is not only measured by their ability to diminish tumor growth without damaging healthy tissues but also by their site specificity and efficient agent delivery, leading to enhanced therapeutic efficacy and negligible adverse effects.One of the methods to develop such NPs is covalent binding, in which relatively stable nanocarriers with both targeting and therapeutic capability are produced.2–4 However, this method is tedious and involves chemical modification. Moreover, if anticancer molecules are tethered covalently to nanocarriers, their therapeutic properties would inevitably become different from their intrinsic ones. Another possible disadvantage of the covalent binding method is that its application decreases the therapeutic efficacy of drugs because of robust chemical bonding between the nanocarriers and the drugs, thus limiting their usefulness.5,6 Compared to covalent binding, noncovalent conjugation that includes physical adsorption, electrostatic interaction, π–π stacking, and coordination chemistry is expected to have a lesser impact on conjugated structures. In addition, noncovalent conjugation has the advantage of easy drug release enabled by the breakage of weak interactions between the drug and the nanocarriers.1,7
As specific examples of its application, covalent conjugation has been widely employed for conjugating folate with NPs,8 polymers,9 lipids,10 and proteins.11 However, only a few studies have been conducted to investigate the noncovalent conjugation of folate. Considering the advantages of noncovalent conjugation, the possibility of conjugating folates with various materials via the noncovalent approach needs to be investigated. Duarte et al.12 successfully conjugated folate on a liposome complex via the noncovalent approach. It is now known that initial functionalization of NPs enables noncovalent conjugation of folate with the NPs. However, complex and time-consuming synthesis routes make it challenging to synthesize bioapplicable NPs through covalent and noncovalent processes. Therefore, it is critical to develop a simple and rapid strategy for synthesizing NPs with medical applications.
We have previously demonstrated a facile phase-transfer strategy for NPs that involved ultrasonic-assisted encapsulation. The NPs process using this strategy showed excellent solubility as well as pH endurance.13 In this study, we expanded this strategy to develop a one-pot approach for not only achieving the phase transfer of NPs in polar solvents but also simultaneously attaching a targeting agent and a therapeutic drug through electrostatic and coordinative interactions, respectively. We chose folate as the cancer-cell-targeting ligand because of its high stability, nonimmunogenic character, low molecular mass (∼441 Da), and high affinity for folate receptors (Kd = ∼10−10 M).14 Further, we selected cis-dichlorodiaminoplatinum (cisplatin, CDDP) as the antitumor drug. We used the one-pot approach for successfully achieving the phase transfer of three types of oil-soluble NPs, namely, AgInS2/ZnS quantum dots (AQDs), gold NPs (GNPs), and Fe3O4 NPs (FNPs), in an aqueous solution. For demonstrating purposes, we conducted another experiment to show that as-prepared AQDs conjugated with folate and CDDP exhibit higher cytotoxicity against tumor cells than QDs carrying only an anticancer drug.
Experimental section
Chemicals
Zinc stearate (90%), 1-dodecanethiol (97%), 1-octadecene (ODE, 90%), potassium ethylxanthate (90%), oleic acid (65–88%), 4′-6-diamidino-2-phenylindole (DAPI, >98%), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 97.5%), linoleic acid (>99%), iron(III) acetylacetonate (Fe(acac)3, 97%), and osmium tetraoxide (4%) were purchased from Sigma-Aldrich (Milwaukee, WI, USA). Zinc chloride (90%) was purchased from Riedel-deHaën AG (Seelze, Germany). Indium acetate (InAc, 99.98%), silver acetate (AgAc, 99%), tetrachloroaurate(III) trihydrate (HAuCl4·3H2O, 99.9%), and glutaraldehyde (25%) were purchased from Alfa-Aesar (Ward Hill, MA, USA). Folate hydrate (>98%) was purchased from T.C.I. Chemical Co. (Tokyo, Japan). Oleylamine (80–90%), cisplatin (CDDP, 99.99%), and benzyl ether (99%) were purchased from Acros Organics (Geel, Belgium). Dimethyl sulfoxide (HPLC grade) was purchased from Scharlau (Barcelona, Spain). All chemicals were used directly without further purification.Synthesis of AQDs
AQDs were synthesized by the solvothermal method according to our previously published procedure.13 AgAc (0.16 mmol), InAc (0.29 mmol), 2.5 mL of 1-dodecanethiol, and 5 mL of ODE were loaded into a reaction vessel equipped with a condenser, a magnetic stirring bar, and a thermometer with an attached Schlenk line. The mixture was stirred vigorously and degassed under vacuum at 60 °C for 30 min. Under argon flow, the resulting solution was subsequently heated to 220 °C for 30 min. ZnS precursors were added drop-wise by means of a syringe pump for 30 min once the temperature reached 220 °C. After the addition was complete, the reaction mixture was cooled to room temperature.Synthesis of GNPs
GNPs were synthesized according to a procedure proposed by Shen et al.15 HAuCl4·3H2O (0.1 mmol) and 5 mL of oleylamine were mixed with 5 mL of toluene in a 50 mL flask. Under argon flow, the mixture was slowly heated to 80 °C under magnetic stirring and kept at this temperature for 6 h. After cooling down the mixture to room temperature, 30 mL of anhydrous ethanol was added to it. The resulting suspension was centrifuged at 4000 rpm (= 1860g-force) for 5 min. The supernatant was discarded, and the obtained GNPs were stored at room temperature.Synthesis of FNPs
FNPs were prepared by reducing an iron solution according to the following reported procedure.16 Fe(acac)3 (3 mmol) was suspended in 15 mL of benzyl ether and 15 mL of oleylamine under argon flow. The mixture was first heated to 110 °C under magnetic stirring and kept at this temperature for 1 h. Subsequently, the temperature was increased to 300 °C for 1 h. Upon heating, the mixture become dark brown in color, indicating the formation of FNPs. After cooling down the mixture to room temperature, the resulting NPs were washed and precipitated by adding ethanol, and were then separated from the supernatant by centrifugation.Phase transfer and surface conjugation of NPs
Ten milligrams of the folate reagent was dissolved in 10 mL of a 2-(N-morpholino) ethanesulfonic acid (MES) buffer solution (pH = 7.4). To that clear solution, 200 μL of oleic acid (0.63 mmol) and 30 μL of 1 M NaOH were gently added. The solution was then vigorously stirred for 1 h until a cloudy solution was obtained. Subsequently, 0.5 mL of hydrophobic AQDs (20 mg in hexane) was added to the resulting solution, and the mixture was ultrasonicated using a high-intensity ultrasonic probe (VCX 130 PB, Sonics and Materials Inc., Newton, CT) operated at 50 Hz with 130 W power until the hexane and aqueous phase were mixed homogenously. After sonication, the resultant mixture was centrifuged at 6000 rpm (=3870g-force) for 10 min to speed-up the separation of the hexane and aqueous phase. The aqueous solution was then collected and passed through a 0.22 μm nylon filter (CHROMAFIL Xtra PA-20/25, Macherey-Nagel & Co., Germany) to remove aggregated NPs. The excess folate was reduced by dialysis performed with DI water using a polyethersulfone membrane (MWCO 3000 Da; Cellu Sep H1, Orange Scientifique, Belgium) for 24 h. The as-prepared AQDs conjugated with folate are hereby referred to as AQD@folate. Similarly, the corresponding folate-conjugated NPs containing GNPs, TNPs, and FNPs are hereafter denoted as GNP@folate, TNP@folate, and FNP@folate, respectively.For the preparation of AQDs conjugated with CDDP (denoted as AQD@CDDP hereafter), the same procedure was employed, except that 6.79 mg of CDDP was used instead of folate. AQDs conjugated with both folate and CDDP, abbreviated as AQD@folate/CDDP hereafter, were also prepared using the same procedure employed for AQD@folate, except that folate and CDDP (6.79 mg) were simultaneously added to 10 mL of MES. ICP analysis was further conducted to determine the concentration of the transferred NPs.
CDDP loading and release evaluation
The amount of CDDP was correlated with the Pt concentration, which was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES). The CDDP-containing samples were heated to remove the solvent. The dried samples were then decomposed by treating them with nitric acid, and their mass was subsequently measured by ICP-AES after dissolving them in DI water. The CDDP loading efficiency (LE) and content (LC) were calculated using the following equations:
 | (1) |
 | (2) |
The CDDP release was evaluated by measuring the Pt concentration in CDDP after passing the samples through a dialysis membrane tubing (MWCO 3500); the test was carried out according to a previously reported procedure.17 1 mL of AQD@folate/CDDP in the dialysis membrane tubing was incubated at 37 °C in 40 mL of a PBS buffer (1.37 M NaCl, 14.7 mM KH2PO4, 27 mM KCl, Na2HPO4; pH 7.4). At a pre-defined time, a 1 mL aliquot was taken and diluted up to 5 mL with 2% nitric acid. Then, the CDDP concentration that was equivalent to the Pt concentration was determined by ICP-AES. The total volume of the PBS buffer was maintained by adding the same volume of fresh PBS buffer and the corrected concentration of released CDDP in accordance with the following equation:
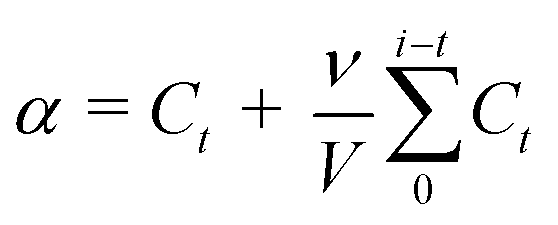 | (3) |
Cell culture
Human cervical (HeLa) and human liver carcinoma (HepG2) cancer cells were cultured in Eagle's Minimum Essential Medium (containing 1.5 g L−1 sodium bicarbonate) supplemented with 1% L-glutamine, 1% antibiotic antimycotic formulation, 1% non-essential amino acid, 1% sodium pyruvate, and 10% fetal bovine serum in a humidified 5% CO2 incubator maintained at 37 °C. The above medium is hereafter referred to as culturing medium.Confocal microscopy study
The HeLa cells and HepG2 were seeded in a 6-well plate in 2 mL of a culturing medium, and were cultured for 24 h. After 1 h of incubation with 300 μL MES buffer containing AQD@folate (1.42 mg mL−1), the cells were washed gently thrice with PBS and then fixed with 75% alcohol for 10 min. Next, the fixed cells were incubated for 17 min at room temperature with 2 mL (0.05 μg mL−1) of DAPI in PBS. After staining, the cells were imaged using a Leica TCS SP2 laser scanning confocal microscope equipped with three lasers—a tunable argon ion laser (458, 488, 514 nm), a green HeNe laser (543 nm), and a red HeNe laser (633 nm)—with three separate photomultiplier tubes for detection.Cytotoxicity study
Cell cytotoxicity against AQDs was investigated by means of an MTT assay. HeLa cells were seeded in a 12-well plate at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well. After 24 h of cell attachment, the cells were washed with PBS and incubated with a specific concentration of free CDDP, AQD@folate, AQD@CDDP, and AQD@folate/CDDP for another 24 h. The cells were then washed twice with PBS and 1 mL of an MTT reagent (0.5 mg mL−1) was then added, followed by incubation for 4 h to allow the formation of the formazan dye. After the medium was carefully removed, 200 mL of dimethyl sulfoxide was subsequently added to dissolve dark-blue formazan crystals. The amount of dark-blue formazan crystals generated by the live cells was proportional to the number of live cells. Absorbance was measured at 570 nm by using a Biotech Powerwave XS plate reader, and calculated according to the equation (Atest/Ablank) × 100%, where A is absorbance.
000 cells per well. After 24 h of cell attachment, the cells were washed with PBS and incubated with a specific concentration of free CDDP, AQD@folate, AQD@CDDP, and AQD@folate/CDDP for another 24 h. The cells were then washed twice with PBS and 1 mL of an MTT reagent (0.5 mg mL−1) was then added, followed by incubation for 4 h to allow the formation of the formazan dye. After the medium was carefully removed, 200 mL of dimethyl sulfoxide was subsequently added to dissolve dark-blue formazan crystals. The amount of dark-blue formazan crystals generated by the live cells was proportional to the number of live cells. Absorbance was measured at 570 nm by using a Biotech Powerwave XS plate reader, and calculated according to the equation (Atest/Ablank) × 100%, where A is absorbance.
Transmission electron microscopy (TEM) imaging of cells
HeLa cells were seeded in a 6-well plate in 2 mL of a culturing medium, and were cultured for 24 h. They were then reincubated for 1 h after the addition of 300 μL of various NPs conjugated with folate. The cells with internalized NPs were trypsinized, centrifuged, and washed with PBS. The resultant cells were then prefixed with 0.2% glutaraldehyde at 4 °C for 5 min and twice with 1% glutaraldehyde at 4 °C for 60 min. They were then post-fixed with 1% osmium tetraoxide for 60 min at room temperature under dark conditions. After washing with 0.1 M PBS, the samples were dehydrated using increasing concentrations of ethanol (50, 70, 80, 90, and 100%) for 15 min. Subsequently, the pellets were embedded in a fresh epoxy resin and polymerized in an oven at 60 °C for 48 h. The epoxy resin, containing 10 g of ERL 4221 (cycloaliphatic epoxide resin ERL 4221), 26 g of nonenyl succinic anhydride (NSA), 6 g of DER-736 epoxy resin (polyglycol dieposides product), and 0.2 g of 2-(dimethylamino) ethanol (DMAE), was introduced gradually into the cell pellets after dehydration. The polymerized blocks were allowed to cool at room temperature before sectioning. Ultrathin sections (∼70 nm thickness) were cut with a diamond knife on an ultramicrotomy apparatus (Leica Microsystems, Germany), and were then transferred to a copper grid.Characterization
Absorption spectra of the samples were measured using a JASCO V-630 spectrometer. Metal concentrations were analyzed using an inductively coupled plasma atomic emission spectrophotometer JY 2000-2 (Jobin Yvon Horiba). Low-magnification TEM images were recorded on a Hitachi H7100 electron microscope operating at 100 kV. High-resolution TEM images were obtained using an FEI Tecnai G2 F20 microscope (Philips, Holland), equipped with a field emission gun operating at an accelerating voltage of 200 kV. The structural properties of the samples were analyzed by X-ray diffraction (XRD) on a Rigaku 18 kW rotating-anode-source X-ray diffractometer with the Cu Kα1 line (λ = 1.54 Å). Fourier transform infrared (FTIR) spectra of the samples were measured using a Bio-Rad FTS-3500 spectrometer. The average hydrodynamic diameter and zeta potential of the samples were measured using a Malvern Nano-ZS 90 dynamic light scattering (DLS) instrument equipped with a 22 mW He–Ne laser operating at λ = 632.8 nm.Results and discussion
Two strategies for achieving the simultaneous phase transfer and surface bioconjugation of NPs via the one-pot approach were adopted (Scheme 1). The first strategy (denoted as (i)) involved hydrophobic interactions and electrostatic protocol; in this strategy, NPS, oleic acid, and folate reagent were incorporated simultaneously. This strategy was utilized to immobilize a folate-targeting agent on the NPs, and this was accompanied by phase transfer. The second strategy (denoted as (ii)) involved hydrophobic as well as electrostatic and coordinative interactions. Strategy (ii) was also a one-pot strategy similar to strategy (i), except that an additional anticancer drug, CDDP, was introduced into the same reaction vessel. This was done to synthesize folate-targeting therapeutic NPs. | ||
| Scheme 1 Schematic illustration of strategies (i) and (ii) based on one-pot approach. Strategy (i) involves simultaneous (a) phase transfer and (b) folate attachment on NPs via electrostatic interactions. Strategy (ii) is used to synthesize folate-targeting therapeutic NPs. | ||
Phase transfer and folate attachment on NPs via strategy (i)
Strategy (i) is based on the general principle of our approach: (a) ultrasonication is performed so that the aliphatic side of oleic acid ligands generate an interdigitated bilayer with hydrophobic capping agents on the surface of various NPs through van der Waals interactions,13,18,19 and a carboxylic moiety from oleic acid becomes exposed on the outermost surface of the NPs; and (b) such resulting NPs with a negatively charged carboxylic moiety would interact electrostatically with nearby positively charged cations present on the pterin rings of the folate reagent. In solution, the introduction of a certain amount of NaOH to adjust pH to 6–7 can enhance electrostatic interactions, and this can result in an improved formation of a well-defined electrostatic double layer. This can be attributed to the fact that the carboxylic groups present in oleic acid are deprotonated (pKa = 4.95) and are thus negatively charged, while the pterin rings of folate are positively charged because of their higher pKa (=7.9).20–22In order to implement proposed strategy (i), we synthesized three types of hydrophobic NPs, as stated in the Introduction: AQDs, GNPs, and FNPs; these NPs are commonly used for conjugation with cancer therapeutic drugs owing to their biocompatibility and low toxicity.23–25 These NPs were chosen because they have been widely employed for biological applications. We subjected these NPs to TEM and XRD analyses; the results are presented in Fig. S1 (ESI†). The conjugation of folate with various NPs was investigated using UV-Vis, FTIR spectroscopy, and dynamic light scattering (DLS), the results of which are shown in Fig. 1. The UV-Vis spectra for free folate molecules showed characteristic absorbance bands at 304 and 390 nm; these bands were attributed to π–π* and n–π* transitions in the pterin ring and enone moiety of folate, respectively.26,27 After conjugation with folate, the UV-Vis spectra of all NPs exhibited a characteristic absorbance band consistently at the same wavelengths of 304 and 390 nm (Fig. 1a, c and e). In contrast, no hydrophobic NPs displayed these characteristic absorbance bands in the wavelength range 250–400 nm. This result proves that folate was successfully conjugated with NPs via the one-pot approach. Further, the results of FTIR analysis revealed that after folate immobilization, most of the characteristic vibrational modes of folate also appeared in the FTIR spectra of the NPs, such as C–H stretching at 2928 and 2843 cm−1, aromatic ring stretching of the pterin ring, and peaks located in the range 1476–1694 cm−1 that correspond to p-amino benzoic acid moieties of folate. The peaks located at 1339 and 916 cm−1 are also evidence of these moieties, corresponding to aromatic C–H in-plane and out-of-plane bending in folate, respectively. The increased intensity of the –OH stretching vibration frequency at 3200 cm−1 also reflects the contribution of folate molecules conjugated with the NPs. Additionally, the DLS results showed that the hydrodynamic diameter of each NP type after conjugation with folate was larger than that of the corresponding original NP type (Fig. 1g). This was mainly attributed to the formation of the interdigitated bilayer, electrostatic layer, and hydration layer surrounding individual NPs after conjugation with folate via the one-pot approach. Since the absorption spectra of folate molecules showed a characteristic absorbance band at 390 nm, we used this wavelength position to track folate concentration, which was absorbed in AQD@folate/CDDP. The absorbed and released folate was determined by comparison with a calibration curve. According to the calibration curve, approximately 15.3%, 19.0%, and 17.8% of the total folate were coupled to the AQDs, GNPs, and FNPs, respectively.
 | ||
| Fig. 1 UV-Vis absorption spectra of AQDs (a), GNPs (c), and FNPs (e) before (red line) and after (blue line) conjugation with folate. The absorption spectra of neat folate (green line) are also shown. Inset: photograph showing NPs dissolved in hexane (left) and water (right). FTIR spectra in the wavelength range 400–4000 cm−1 of AQDs (b), GNPs (d), and FNPs (f). (g) Hydrodynamic size distribution graphs for AQDs, GNPs, and FNPs before (red bar) and after (violet bar) conjugation with folate. | ||
The three-dimensional confocal imaging of AQD@folate-incubated HeLa cells was also performed at different depths along the z-axis (Fig. S3†). Results of this analysis showed that AQD@folate was distributed in the HeLa cells in multiple cross-sections. The results also indicated successful internalization of AQD@folate into the cells instead of only aggregation on the cell surface. For comparison, control experiments were performed with AQDs lacking folate conjugations (AQD@OA); no noticeable intracellular quantum dot fluorescence was observed (Fig. 2b and S4a†). These confocal images also revealed that endocytosis of AQD@folate on the Hela cells was mediated by folate receptors that were abundant on the cell membrane. We predicted that the cellular uptake of AQD@folate was affected by folate receptor mediation. To prove this, the confocal image of a Hela cell treated with AQD@folate by incubation for 1 h at 4 °C was taken (Fig. 3). As explained in several reports,28–30 endocytosis on a cell can be completely prevented at low temperatures. Nevertheless, a folate–attributed material has to change to reach a cell membrane. As a result, the confocal image exhibits yellow fluorescence around the Hela cells. To further confirm the targeting effect of AQD@folate, we also used HepG2 cell as a negative control since several groups have reported that HepG2 cell is a folate receptor deficient cancer cell line.31–33 HepG2 cell was treated with AQD@folate under similar conditions. Fig. S4b† shows that only a weak yellow emission in HepG2 cells could be observed. This result suggests that HepG2 cell is incapable of internalizing significant amounts of AQD@folate because HepG2 cell rarely has folate receptors on the membrane surface. Furthermore, ultrastructural observations of the cells incubated with GNP@folate and FNP@folate were recorded using TEM (Fig. 2c, d, S5 and S6†). In Fig. 2c, GNP@folate appear as black dots scattered and randomly spread inside the cell cytoplasm, suggesting that GNP@folate were endocytosed by the HeLa cells. Similarly, FNP@folate also appeared inside the cell cytoplasm, as shown in Fig. 2d. Therefore, after folate-receptor-mediated endocytosis, the uptaken GNP@folate and FNP@folate exhibited good dispersion and retained their spherical shape and uniform size after internalization into cells.
 | ||
| Fig. 2 (a) Confocal images of HeLa cells treated for 1 h with AQD@folate, as compiled from transmission and luminescence images of DAPI (blue) and AQD@folate (yellow). (b) Similar confocal images of HeLa cells treated for 1 h with AQD@OA and DAPI. Scale bars represent 40 μm. Transmission electron micrographs of (c) GNP@folate and (d) FNP@folate located on HeLa cells after incubation for 1 h. | ||
 | ||
| Fig. 3 Confocal laser images of HeLa cells treated with AQD@folate by incubation for 1 h at 4 °C: (a) transmission image, (b) yellow emission by AQD@folate, and (c) overlapping of images shown in (a) and (b). Scale bars represent 20 μm. | ||
The suitability of designed NP@folate for biomedical applications can be reflected by its stability against various pH and ionic strength values. We chose AQD@folate as representative NPs for colloidal stability analysis. Photographs of AQD@folate with pH in range 3–12 at room temperature are shown in Fig. S7 (ESI†). It can be seen that no significant change occurred in AQD@folate until 24 h for solutions with pH above 5. Even at low pH, the solution color became strong yellow, owing to the pterin ring conformation of folate; the actual destruction of AQD@folate occurred at pH 3 and 4. Hereafter, a neutral pH solution was used, and AQD@folate maintained its structure for 24 h against NaCl concentration up to 0.5 M. These results revealed that AQD@folate are tolerant to physiological conditions (most of the acid in the intracellular compartment at pH 5; most of the base in the pancreas region at pH 8.1; human salt concentration: 0.15 M).34
CDDP Loading with Strategy (ii)
For achieving NPs with targeting and therapeutic capability, CDDP was used as a model anticancer drug. CDDP has been used in cancer chemotherapy ever since its approval for the treatment of testicular cancer.35 At present, it is one of the most widely used anticancer drugs, and its binding to DNA causes a significant distortion in the double-helix structure that acts as a recognition signal in remedy and other cellular processes.36 Our second strategy (ii) is based on the fact that CDDP easily forms a complex with nanomaterials through coordination of the platinum atom to terminal carboxylate groups on the surface of the nanomaterials; this has been well documented in the literature.37–42 Therefore, active targeting NPs with therapeutic capability can be formed through the combined synergistic effects of the consecutive adsorption of an oppositely charged reagent and coordinative interactions of the platinum atom with carboxylic acid ligands. This implies that a carboxylic moiety of an oleic acid ligand exposed on the outermost surface of a negatively charged NP is conjugated to a folate-targeting reagent via electrostatic interactions, while a folate molecule is coordinated to the platinum atom of CDDP through the coordination capability of the α- and γ-carboxylate groups present on glutamic acid in folate. Additionally, two available carboxylate groups of adjacent oleic acid ligands on the surface of a NP are also capable of coordinating with the platinum atom to form a complex by substituting the two chloride ligands in CDDP. With this strategy, the yield of the resulting NPs was around 65.4%, 57.4%, and 57.8% for AQD@folate/CDDP, GNP@folate/CDDP, and FNP@folate/CDDP, respectively.Zeta potential measurements demonstrated a clear response corresponding to successful conjugation via strategy (ii). As shown in Table 1, zeta potential revealed that all the NPs were negatively charged and showed the highest zeta potential values in the co-presence of folate and CDDP; moreover, they showed the lowest zeta potential values in the presence of only the folate reagent. For example, initially, the zeta potential of purified AQD@OA was −67.17 mV in an aqueous suspension, suggesting that AQD@OA were stabilized by the outer layers of ionized carboxylic groups. Notably, the zeta potential values of AQD@folate became more negative (−89.07 mV) owing to the numerous carboxyl moieties on the surface of AQD@folate because each folate reagent molecule contains α- and γ-carboxyl groups. However, the zeta potential of AQD@folate/CDDP was measured to be around −56.6 mV. These results suggest that the amine group of CDDP was the outermost site on the NP, and also indicate that successful conjugation of AQDs with folate and CDDP had occurred.
| Sample | Zeta Potential (mV) | LE (%) | LC (%) |
|---|---|---|---|
| AQD@OA | −67.17 ± 1.64 | — | — |
| AQD@folate | −89.07 ± 6.99 | — | — |
| AQD@CDDP | −10.00 ± 8.25 | 85.86 ± 1.92 | 9.05 ± 0.25 |
| AQD@folate/CDDP | −56.60 ± 6.30 | 79.49 ± 2.45 | 3.36 ± 0.03 |
| GNP@OA | −55.4 ± 2.60 | — | — |
| GNP@folate | −72.8 ± 1.60 | — | — |
| GNP@CDDP | −51.40 ± 1.44 | 48.62 ± 0.32 | 5.83 ± 0.04 |
| GNP@folate/CDDP | −61.20 ± 1.18 | 59.54 ± 0.78 | 4.70 ± 006 |
| FNP@OA | −29.00 ± 7.30 | — | — |
| FNP@folate | −90.90 ± 2.10 | — | — |
| FNP@CDDP | −42.70 ± 0.26 | 68.81 ± 0.27 | 9.17 ± 0.04 |
| FNP@folate/CDDP | −55.13 ± 2.85 | 70.14 ± 0.71 | 4.29 ± 0.05 |
Associating CDDP on NPs slightly reflects the drug delivery potential of designed NPs. However, quantitative analysis of CDDP must be carried to ascertain the effectiveness of the strategy employed in the drug delivery process. The LE and LC values of different NPs are presented in Table 1. LE values for AQD@folate/CDDP, GNP@folate/CDDP, and FNP@folate/CDDP are 79.49 ± 2.45%, 59.54 ± 0.78%, and 70.14 ± 0.71%, respectively, whereas LC values are 3.36 ± 0.03%, 4.70 ± 006%, and 4.29 ± 0.05%, respectively. On the basis of results obtained in previous studies, i.e., CDDP loading content in the range 3.0–88.3%,43–45 the low LC results obtained by employing the present strategy are competitively acceptable. Moreover, LE values for AQD@CDDP, GNP@CDDP, and FNP@CDDP were calculated to be 85.86 ± 1.92%, 48.62 ± 0.32%, and 68.81 ± 0.27% respectively. These results suggest the possibility of direct coordinative interaction between CDDP and various NP@OA.
CDDP released and therapeutic capability of resultant AQDs
A comparison of the kinetic release profiles of CDDP from AQD@folate/CDDP in PBS with specific pH values at 37 °C is shown in Fig. 4a. The figure clearly shows that CDDP release was pH dependent. Decreasing pH will break CDDP coordination, resulting in its release from the NPs. Preceded by burst release during the first 24 h, CDDP diffusion at pH 5 increased with the release rate at t1/2 = 27.91 h. However, the complete release of free CDDP in the initial stage indicated that AQD@folate/CDDP showed the ability of controlled release. This implies possible interaction between CDDP and folate on the AQD surface. Moreover, a similar procedure was used to quantify the amount of released folate after passing the samples through a dialysis membrane tubing. Fig. 4a shows that the release of folate from AQD@folate/CDDP was pH-independent. This is possibly because folate, carrying both positively and negatively charged functional groups, is capable of providing much better stability at various pH values. | ||
| Fig. 4 (a) Release of AQD@folate/CDDP (solid lines) and folate (dash lines) incubated in PBS at pH 7.4 (red) and 5 (green). (b) In vitro viability of HeLa cells incubated with free folate (blue), AQD@OA (green), AQD@folate (yellow), and AQD@folate/CDDP (red) at different concentrations. Deionized water (0 μg mL−1) served as the control. Error bars in the graph represent standard deviations (n = 3). | ||
Furthermore, we consistently used AQDs as representative NPs for detailed discussion on the enabling of targeting and therapeutic capability. Several groups have recently reported the use of quantum dots for simultaneous imaging and therapeutic applications that allow monitoring of the position of targeted tissue regions.46–51 To investigate the effect of the potential toxicity of free folate, AQD@OA, AQD@folate, and AQD@folate/CDDP on cell viability, an MTT assay was performed on HeLa cells for 24 h (Fig. 4b). The results exhibited that free folate, AQD@OA, and AQD@folate were non-toxic for Hela cells, viability was over 80% even when the concentration was increased to 400 μg mL−1, and the NPs gained toxicity properties after the association of CDDP. These results imply that the toxicity of the designed NPs can be attributed to CDDP. The cytotoxic effect of AQD@folate/CDDP against HepG2 cancer cells was also evaluated in vitro via the MTT assay (Fig. S8†). It was observed that the viability of HepG2 is higher than that of HeLa cancer cells under identical AQD@folate/CDDP concentrations. This may be attributed to the fact that the AQD@folate/CDDP preferentially targeted HeLa cells via receptor mediated targeting and thus enhanced cellular uptake of the drug.
Toxicity analysis was conducted to demonstrate that the CDDP uptake on the HeLa cells by comparing the viability of the cells incubated with AQD@folate/CDDP, AQD@CDDP, and free CDDP at seven different Pt concentrations in the range 1–40 μg mL−1 (Fig. 5a). The Pt concentration in the solution was determined by ICP-AES. After 24 h of incubation, significant cytotoxicity was observed in the HeLa cells. It was also found that the cytotoxicity of AQD@folate/CDDP was higher than that of AQD@CDDP at all tested concentrations. For example, 49.8% of HeLa cells were retained when treated with AQD@CDDP containing 9.1 μg mL−1 of Pt, but only around 26% cells were viable when exposed to AQD@folate/CDDP with the same Pt dose. For comparison, HeLa cells were also treated with free CDDP at the same concentrations. Fig. 5a shows that free CDDP exhibited strong inhibition of cell proliferation at low concentrations (i.e., 20% viability at 1 μg mL−1), while AQD@CDDP and AQD@folate/CDDP exhibited dose-dependent cytotoxicity for HeLa cells. We then measured the IC50 values of AQD@folate/CDDP, AQD@CDDP, and free CDDP (Shown on Fig. S9†) to be 1.477 μg mL−1, 2.856 μg mL−1, and 0.272 μg mL−1, respectively. As compared to the results for AQD@CDDP, this result suggests that AQD@folate/CDDP could provide more assistance to delivery of CDDP into tumor cells with greater cytotoxicity because of folate-receptor-mediated cellular uptake. The higher cytotoxicity of free CDDP than that of other samples might be partially attributed to the intrinsically low molecular weight of CDDP, which results in fast diffusion of CDDP into HeLa cells. However, its low molecular weight also reflects a short blood circulation time when administered intravenously, which is a frequently used route for targeted drug delivery. In addition, we investigated the efficient toxicity of HeLa cells at low CDDP doses. Fig. 5b shows a comparison of cell viability after treatment with low CDDP dose (1 μg mL−1 of Pt concentration) as a function of time. The results exhibit similar patterns with decreasing cell viability and increasing time. The figure also shows that delay times are needed for CDDP release and that AQD@folate/CDDP showed toxicity even at low CDDP doses.
 | ||
| Fig. 5 (a) In vitro viability of HeLa cells incubated with (i) AQD@CDDP, (ii) AQD@folate/CDDP, and (iii) free CDDP as a function of Pt concentration. Deionized water (0 μg mL−1) served as the control. (b) Improved in vitro viability of HeLa cells incubated with deionized water as control (blue) and same Pt concentration of CDDP (0.33 μg mL−1) from AQD@folate/CDDP (purple), AQD@CDDP (magenta), and free CDDP (yellow) as a function of time. All error bars in the graph represent standard deviations (n = 3). *p < 0.1, **p < 0.05. | ||
Conclusions
We devised a facile strategy for enabling NPs with simultaneous phase transfer and noncovalent surface bioconjugation via a one-pot approach. This approach was demonstrated for three types of NPs, namely, AQDs, GNPs, and FNPs. Colloidal stability of these NPs under physiological conditions and CDDP loading results revealed that the proposed strategy is an excellent way to rapidly obtain simple and high-efficiency bioapplicable NPs. Confocal laser scanning microscopy revealed that AQD@folate were taken up effectively by HeLa cells and an MTT assay demonstrated that they exhibited negligible cytotoxicity after 24 h in HeLa cells (>80% cell viability at concentrations up to 400 μg mL−1). In contrast, after conjugation with CDDP, AQDs can be used as a tumor-targeting therapeutic probe that would exhibit efficient cytotoxicity for HeLa cells even at low doses of CDDP. Given the simplicity of the proposed strategy, we anticipate that many other hydrophobic NPs can be transformed into targeting therapeutic NPs by selecting appropriate reagents.Acknowledgements
The authors thank the Ministry of Science and Technology of the Republic of China for financially supporting this research under Contract no. 102-2628-M-011-001-MY3.Notes and references
- Y. Pan, H. Bao, N. G. Sahoo, T. Wu and L. Li, Adv. Funct. Mater., 2011, 21, 2754–2763 CrossRef CAS.
- Y. Chang, N. Liu, L. Chen, X. Meng, Y. Liu, Y. Li and J. Wang, J. Mater. Chem., 2012, 22, 9594–9601 RSC.
- F. Wang, G. M. Pauletti, J. Wang, J. Zhang, R. C. Ewing, Y. Wang and D. Shi, Adv. Mater., 2013, 25, 3485–3489 CrossRef CAS PubMed.
- X. Yang, H. Hong, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, Y. Xiao, Y. Yang, Y. Zhang, R. J. Nickles, W. Cai, D. A. Steeber and S. Gong, Biomaterials, 2011, 32, 4151–4160 CrossRef CAS PubMed.
- B. R. Liu, Y.-W. Huang, J. G. Winiarz, H.-J. Chiang and H.-J. Lee, Biomaterials, 2011, 32, 3520–3537 CrossRef CAS PubMed.
- F. Sousa, S. Mandal, C. Garrovo, A. Astolfo, A. Bonifacio, D. Latawiec, R. H. Menk, F. Arfelli, S. Huewel, G. Legname, H.-J. Galla and S. Krol, Nanoscale, 2010, 2, 2826–2834 RSC.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- S. Ge, F. Liu, W. Liu, M. Yan, X. Song and J. Yu, Chem. Commun., 2014, 50, 475–477 RSC.
- Y. Liu, K. Li, J. Pan, B. Liu and S.-S. Feng, Biomaterials, 2010, 31, 330–338 CrossRef CAS PubMed.
- R. Hood, C. Shao, D. Omiatek, W. Vreeland and D. DeVoe, Pharm. Res., 2013, 30, 1597–1607 CrossRef CAS PubMed.
- Y. Ren, S. M. Wong and L.-Y. Lim, Bioconjugate Chem., 2007, 18, 836–843 CrossRef CAS PubMed.
- S. Duarte, H. Faneca and M. C. Pedroso de Lima, J. Controlled Release, 2011, 149, 264–272 CrossRef CAS PubMed.
- M. Z. Fahmi and J.-Y. Chang, Nanoscale, 2013, 5, 1517–1528 RSC.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2007, 41, 120–129 CrossRef PubMed.
- C. Shen, C. Hui, T. Yang, C. Xiao, J. Tian, L. Bao, S. Chen, H. Ding and H. Gao, Chem. Mater., 2008, 20, 6939–6944 CrossRef CAS.
- Z. Xu, C. Shen, Y. Hou, H. Gao and S. Sun, Chem. Mater., 2009, 21, 1778–1780 CrossRef CAS.
- K. Cheng, S. Peng, C. Xu and S. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS PubMed.
- J.-Y. Chang, J.-M. Lin, L.-F. Su and C.-F. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 8740–8752 CAS.
- A. Prakash, H. Zhu, C. J. Jones, D. N. Benoit, A. Z. Ellsworth, E. L. Bryant and V. L. Colvin, ACS Nano, 2009, 3, 2139–2146 CrossRef CAS PubMed.
- V. D. Monópoli, A. H. Thomas and A. L. Capparelli, Int. J. Chem. Kinet., 2000, 32, 231–237 CrossRef.
- J. D. M. Patring, S. A. Lanina and J. A. Jastrebova, J. Sep. Sci., 2006, 29, 889–904 CrossRef CAS.
- R. J. Pugh, Colloids Surf., 1986, 18, 19–41 CrossRef CAS.
- C. K. Kim, P. Ghosh, C. Pagliuca, Z.-J. Zhu, S. Menichetti and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 1360–1361 CrossRef CAS PubMed.
- L. Liu, H. Rui, I. Roy, G. Lin, L. Ye, J. L. Reynolds, J. Liu, J. Liu, S. A. Schwartz, X. Zhang and K.-T. Yong, Theranostics, 2013, 3, 109–115 CrossRef CAS PubMed.
- M. K. Yu, Y. Y. Jeong, J. Park, S. Park, J. W. Kim, J. J. Min, K. Kim and S. Jon, Angew. Chem., Int. Ed., 2008, 47, 5362–5365 CrossRef CAS PubMed.
- J. Ma, P. Huang, M. He, L. Pan, Z. Zhou, L. Feng, G. Gao and D. Cui, J. Phys. Chem. B, 2012, 116, 14062–14070 CrossRef CAS PubMed.
- B. Sahoo, K. S. P. Devi, S. K. Sahu, S. Nayak, T. K. Maiti, D. Dhara and P. Pramanik, Biomater. Sci., 2013, 1, 647–657 RSC.
- J. K. Jaiswal, H. Mattoussi, J. M. Mauro and S. M. Simon, Nat. Biotechnol., 2003, 21, 47–51 CrossRef CAS PubMed.
- P. Jiaqi, W. Jumeng, S. Jianmin, G. Shikuan, S. Yingzhuo, Z. Xin and F. Boxue, J. Phys. D: Appl. Phys., 2014, 47, 045504 CrossRef.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- J. Dai, S. Zou, Y. Pei, D. Cheng, H. Ai and X. Shuai, Biomaterials, 2011, 32, 1694–1705 CrossRef CAS PubMed.
- M. Wang, H. Hu, Y. Sun, L. Qiu, J. Zhang, G. Guan, X. Zhao, M. Qiao, L. Cheng, L. Cheng and D. Chen, Biomaterials, 2013, 34, 10120–10132 CrossRef CAS PubMed.
- H. Cheng, J.-L. Zhu, X. Zeng, Y. Jing, X.-Z. Zhang and R.-X. Zhuo, Bioconjugate Chem., 2009, 20, 481–487 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- C. X. Zhang, P. V. Chang and S. J. Lippard, J. Am. Chem. Soc., 2004, 126, 6536–6537 CrossRef CAS PubMed.
- W. Ding, M. Wada, H. Minamikawa, N. Kameta, M. Masuda and T. Shimizu, Chem. Commun., 2012, 48, 8625–8627 RSC.
- J. Gu, J. Liu, Y. Li, W. Zhao and J. Shi, Langmuir, 2012, 29, 403–410 CrossRef PubMed.
- J. Gu, S. Su, Y. Li, Q. He, J. Zhong and J. Shi, J. Phys. Chem. Lett., 2010, 1, 3446–3450 CrossRef CAS.
- R. Xing, X. Wang, C. Zhang, J. Wang, Y. Zhang, Y. Song and Z. Guo, J. Mater. Chem., 2011, 21, 11142–11149 RSC.
- C. Xu, B. Wang and S. Sun, J. Am. Chem. Soc., 2009, 131, 4216–4217 CrossRef CAS PubMed.
- W. Zhu, Y. Li, L. Liu, Y. Chen, C. Wang and F. Xi, Biomacromolecules, 2010, 11, 3086–3092 CrossRef CAS PubMed.
- N. Alam, V. Khare, R. Dubey, A. Saneja, M. Kushwaha, G. Singh, N. Sharma, B. Chandan and P. N. Gupta, Mater. Sci. Eng., C, 2014, 38, 85–93 CrossRef CAS PubMed.
- D. Ding, Z. Zhu, Q. Liu, J. Wang, Y. Hu, X. Jiang and B. Liu, Eur. J. Pharm. Biopharm., 2011, 79, 142–149 CrossRef CAS PubMed.
- X. Li, R. Li, X. Qian, Y. Ding, Y. Tu, R. Guo, Y. Hu, X. Jiang, W. Guo and B. Liu, Eur. J. Pharm. Biopharm., 2008, 70, 726–734 CrossRef CAS PubMed.
- V. Bagalkot, L. Zhang, E. Levy-Nissenbaum, S. Jon, P. W. Kantoff, R. Langer and O. C. Farokhzad, Nano Lett., 2007, 7, 3065–3070 CrossRef CAS PubMed.
- Y. Choi, M. Kim, Y. Cho, E. Yun and R. Song, Nanotechnology, 2013, 24, 075101 CrossRef PubMed.
- T. Jamieson, R. Bakhshi, D. Petrova, R. Pocock, M. Imani and A. M. Seifalian, Biomaterials, 2007, 28, 4717–4732 CrossRef CAS PubMed.
- Z. Liu, Q. Lin, Q. Huang, H. Liu, C. Bao, W. Zhang, X. Zhong and L. Zhu, Chem. Commun., 2011, 47, 1482–1484 RSC.
- Y. Zhou, L. Shi, Q. Li, H. Jiang, G. Lv, J. Zhao, C. Wu, M. Selke and X. Wang, Biomaterials, 2010, 31, 4958–4963 CrossRef CAS PubMed.
- P. Zrazhevskiy, M. Sena and X. Gao, Chem. Soc. Rev., 2010, 39, 4326–4354 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11582a |
| This journal is © The Royal Society of Chemistry 2014 |