
An eco-friendly synthesis of novel 3,5-disubstituted-1,2-isoxazoles in PEG-400, employing the Et3N-promoted hydroamination of symmetric and unsymmetric 1,3-diyne-indole derivatives†
Abstract
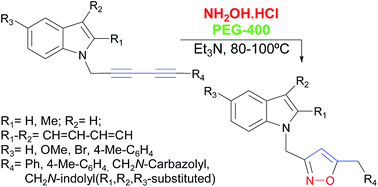
A facile, efficient and atom-economic synthesis of 3,5-disubstituted 1,2-isoxazoles bearing indole moieties, is reported. The synthesis of these isoxazoles was carried out by the triethylamine-promoted reaction of symmetric and unsymmetric 1,3-diyne indole derivatives with hydroxylamine in PEG-400, as an eco-friendly solvent, under relatively mild conditions. The synthesis of the starting 1,3-diyne indole derivatives was performed by the aerobic self-coupling of diversely functionalized N-propargyl indoles and N-propargyl carbazole under copper catalysis, or by the reaction of the propargyl derivatives with phenyl- or p-tolyl-acetylene under combined nickel and copper catalysis. The isoxazolation reaction was optimized, its scope and limitations were studied and a detailed reaction mechanism was proposed.
 Please wait while we load your content...
Please wait while we load your content...

