
A metal-free and a solvent-free synthesis of thio-amides and amides: an efficient Friedel–Crafts arylation of isothiocyanates and isocyanates†
Abstract
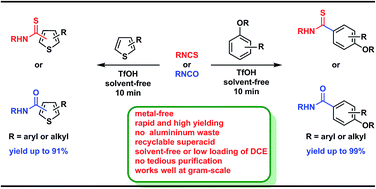
A rapid, metal-free and solvent-free (very low loading of solvent in few cases) reaction conditions for synthesizing thioamides and amides using a Bronsted super acid such as triflic acid has been developed. This method shows a broad substrate scope with a variety of electron-rich arenes including thiophene derivatives. The reaction works well for both aromatic as well as aliphatic isothiocyanates. Most of the thioamides are obtained in excellent yields in short reaction times and in most of the examples, a simple work up procedure has been developed which does not require further purification.
 Please wait while we load your content...
Please wait while we load your content...

