
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The selective conversion of D-limonene to p,α-dimethylstyrene†
S. A.
Sanchez-Vazquez
,
T. D.
Sheppard
,
J. R. G.
Evans
and
H. C.
Hailes
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: h.c.hailes@ucl.ac.uk; Fax: +44 (0)20 7679 7463
First published on 10th November 2014
Abstract
Reaction conditions to facilitate the conversion of D-limonene selectively to p,α-dimethylstyrene (DMS) are described, in order to subsequently produce polymeric materials from biomass sourced from food waste. Limonene was dehydrogenated with several palladium catalysts and different solvents and bases, with copper chloride as oxidant at temperatures of 70–120 °C. Reaction conditions were identified using Pd(OAc)2 for the selective formation of only DMS from limonene in 2–5 hours, enabling the facile separation of DMS from unreacted starting material by vacuum distillation.
Introduction
D-Limonene 1 is the major constituent of citrus peel oil (up to 90% of the monoterpenes present) and a renewable feedstock from citrus peel waste, which has production levels of approximately 12 Tg per annum.1 Several studies have explored the conversion of limonene into p-cymene 2 and other compounds using for example supported Pd-dehydrogenation catalysts, zeolite supported Pd-catalysts (Ce-promoted), heteropolyacid H5PMo10V2O40, silica–alumina supports, and acid activated clays.2–8 However, product selectivity can be a problem, for example when using acid-activated bentonite p-cymene was formed in about 15% yield together with polymeric compounds and non-volatile products,7 although some reaction conditions do lead to high yields.3,4 In other experiments, intermediates and isomers were formed including p-menthene and terpenoline 3.2 Interest in the formation of p-cymene from sustainable feedstocks reflects its importance in many applications including use as a solvent or fragrance ingredient and as an intermediate in fine chemical synthesis.9 Further dehydrogenation of p-cymene produces 1-methyl-4-(2-propenyl)benzene, p,α-dimethylstyrene 4 (DMS), which can be polymerized or copolymerized.10,11The direct conversion of limonene 1 into DMS 4, is particularly attractive, and a recent report by Jaekel and co-workers has described the use of a catalytic heterogeneous method using palladium(II) trifluoroacetate (Pd(OTFA)2) and anhydrous copper chloride (CuCl2) as an oxidant in the presence of 2,6-di-tert-butylpyridine (2,6-tBu2Py) as a sterically hindered base.12 This built upon previous reports describing the use of palladium(II) in endocyclic alkene dehydrogenations, with either maleic acid as a hydride acceptor, or dioxygen as an oxidant.13,14 Studies established that a heterogeneous catalytic system operated to generate DMS 4 in 65% yield (by GC) with a selectivity in favour of DMS 4 [14.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 4:(2 + 3)] (Scheme 1). The amount of the other products 5 and 6 formed was not indicated (Table 1, entry 1), and it is unclear whether the reported yield refers to DMS alone, or the total amount of DMS and 2 and 3.12 Regardless, the formation of DMS alongside these other compounds makes the separation procedure more complex, and the successful purification of DMS was not described.
:1 ratio of 4:(2 + 3)] (Scheme 1). The amount of the other products 5 and 6 formed was not indicated (Table 1, entry 1), and it is unclear whether the reported yield refers to DMS alone, or the total amount of DMS and 2 and 3.12 Regardless, the formation of DMS alongside these other compounds makes the separation procedure more complex, and the successful purification of DMS was not described.
 | ||
| Scheme 1 Conversion of D-limonene to DMS 4 and co-products 2, 3, 5 and 6 (Jaekel et al.).12 | ||
| Entry | Baseb | Total conversion (%) | DMS 4 (%) | p-Cymene 2 (%) | Terpinolene 3 (%) | γ-Terpinene 5 (%) | α-Terpinene 6 (%) |
|---|---|---|---|---|---|---|---|
| a Reactions were carried out in anhydrous DMF at 80 °C (unless indicated) for 40 h using Pd(OTFA)2 (5 mol%), CuCl2 (2 equiv.), 2,6-tBu2Py unless indicated otherwise (3 equiv.). Product ratios determined by GC analysis. b Referred to in (ref. 12) as ‘additive’. c Results from ref. 12. d Non-anhydrous DMF used. e Reaction performed under Ar. f Activated molecular sieves 4 Å added. g Reaction carried out at 120 °C. | |||||||
| 1c,d | 2,6-tBu2Py | 74.8 | 65.4 | 14.3:1, 4:2 + 3 |
14.3:1, 4:2 + 3 |
not given | not given |
| 2d,e,f | 2,6-tBu2Py | 55 | 20 | 0 | 0 | 17 | 18 |
| 3e | 2,6-tBu2Py anhyd. | 70 | 10 | 17 | 8 | 14 | 21 |
| 4e,f | 2,6-tBu2Py anhyd. | 72 | 57 | 1 | 8 | 1 | 5 |
| 5e,f | — | 39 | 11 | 0 | 26 | 0 | 2 |
| 6e,f | 2,6-Lutidine anhyd. | 1.4 | 1.4 | 0 | 0 | 0 | 0 |
| 7e,f,g | 2,6-tBu2Py anhyd. | 98 | 1 | 36 | 34 | 2 | 25 |
| 8e,f,g | 2,6-Lutidine anhyd. | 18 | 18 | 0 | 0 | 0 | 0 |
| 9e,g | 2,6-Lutidine anhyd. | 22 | 22 | 0 | 0 | 0 | 0 |
We are interested in the synthesis of hydrophobic polymers from food wastes, in particular the use of limonene to generate DMS 4, a strategy intended to reduce raw materials costs as the mineral oil price increases, while removing the conflict over land-use between biomass and food production. One problem to overcome however is that the conversion to DMS needs to be highly selective to achieve a facile separation from any remaining limonene: i.e. the formation of side-products with similar physical properties (b.p. at 760 mm Hg: 2 176–178 °C, 3 183–185 °C, 4 186–189 °C, 5 182 °C, 6 173 °C) must be avoided. Here the successful conversion of limonene selectively into DMS for subsequent polymerization chemistries has been investigated.
Results and discussion
Initial reaction conditions and use of alternative bases
In the previous report on the conversion of 1 to 4 and other compounds, the purity of the D-limonene used was not stipulated.12 Nevertheless, in our applications limonene of 97% purity was used (impurities include myrcene, α-pinene), which was felt to be representative of the quality considered for use on a larger scale for DMS production for polymer synthesis. In addition, this was not dried and further purified before use as previously indicated.12 Using the reported conditions (Scheme 1: 2 equiv. CuCl2; 3 equiv. 2,6-tBu2Py) but on a larger scale (6-fold), under an argon atmosphere in DMF (not anhydrous but with the addition of activated 4 Å molecular sieves), although a conversion yield of 55% was noted the selectivity was poor, 4:7 for 4:(5 + 6) (Table 1, entry 2). The reaction was repeated using anhydrous DMF and base, but no molecular sieves (Table 1, entry 3), however 6 became the major product with poor selectivity towards DMS. Using activated molecular sieves, and anhydrous solvent and base, the product distribution and yield were closer to that previously reported, with DMS as the major product with a selectivity of 4:1 DMS 4:(2 + 3 + 5 + 6), and 6:1 4:(2 + 3) (Table 1, entry 4).12 This highlighted the sensitivity of the reaction conditions to the presence of water, indeed when adding water instead of base, the dehydrogenation of limonene was completely inhibited. When base was excluded from the reaction the major product was terpinolene 3 (Table 1, entry 5), which is consistent with the proposed mechanism by Jaekel and co-workers.12 A screen of several non-coordinating hindered bases (N,N-diisopropylethylamine, 2,2,6,6-tetramethylpiperidine, 2,4,6-trimethylpyridine and 2,6-lutidine) highlighted that only the pyridines generated DMS as a single dehydrogenation product (1% yield; Table 1, entry 6 for lutidine result) and due to its lower cost 2,6-lutidine was selected for further experiments.
With a view to increasing the amount of DMS produced using 2,6-tBu2Py the reaction temperature was increased to 120 °C, which gave improved conversion yields (98%) but very little DMS was formed and the major product was p-cymene 2 (Table 1, entry 7). When using 2,6-lutidine however at 120 °C, either in the presence or absence of molecular sieves, DMS 4 was formed as the only product in approximately 20% yield (Table 1, entries 8 and 9), and so these conditions were used in subsequent reactions. Although this yield was not high the reaction selectivity was excellent and will have a major impact on the ease of product purification. These results are also interesting because in previous work using Pd(OTFA)2 in dehydrogenation reactions of cyclohexenes, attempts to neutralise the acid formed using 2,6-lutidine led to a total inhibition of the reaction.13
The use of different solvents and catalysts
The use of alternative solvents were then investigated.15 In cyclopentyl methyl ether (CPME) only p-cymene 2 was formed as a single product in 30% yield (Table 2, entry 1).| Entry | Temp. (°C) | Solvent | Catalyst | Total conversion (%) | DMS 4 (%) | Other products |
|---|---|---|---|---|---|---|
| a Reactions were carried out using the catalyst, solvent and temperature indicated for 40 h with CuCl2 (2 equiv.), 2,6-lutidine (3 equiv.) under argon, unless otherwise indicated. Product ratios determined by GC analysis. b Molecular sieves 4 Å added. c 12 h reaction. d Reaction performed under an oxygen atmosphere without CuCl2. e 3 h with CuCl2 (4 equiv.), 2,6-lutidine (9 equiv.) under argon. CPME: cyclopentyl methyl ether. | ||||||
| 1b | 90 | CPME | Pd(OTFA)2 5 mol% | 30 | 0 | 30% 2 |
| 2b | 70 | 2-MeTHF anhyd. | Pd(OTFA)2 5 mol% | 3 | 0 | 1% 3, 2% 6 |
| 3 | 70 | 2-MeTHF anhyd. | Pd(OTFA)2 5 mol% | 55 | 0 | 25% 2, 30% 6 |
| 4c | 80 | CH3CN anhyd. | PdCl2 5 mol% | 7 | 7 | 0 |
| 5b | 80 | DMF anhyd. | Pd(OAc)2 5 mol% | 2 | 2 | 0 |
| 6 | 120 | DMF anhyd. | Pd(OAc)2 5 mol% | 19 | 19 | 0 |
| 7 | 120 | DMF anhyd. | Pd(OAc)2 2 mol% | 6 | 5 | 1% 5 |
| 8 | 120 | DMF anhyd. | Pd(OAc)2 10 mol% | 0 | 0 | 0 |
| 9c,d | 120 | DMF anhyd. | Pd(OAc)2 5 mol% | 11 | 11 | 0 |
| 10e | 120 | DMF anhyd. | Pd(OAc)2 10 mol% | 39 | 39 | 0 |
Another solvent with sound green credentials is 2-methyl THF (2-MeTHF).15 When used in the presence of molecular sieves only small amounts of 3 and 6 were formed, however, in the absence of molecular sieves 2 and 6 were formed in a much higher combined yield of 55% (Table 2, entries 2 and 3), reflecting problems with the second proposed dehydrogenation step under these conditions.12 The use of heptane resulted in no conversion, presumably due to the poor stability of charged intermediates in this apolar solvent. Alternative lower cost catalysts were then investigated. Palladium chloride (PdCl2) when used with 2,6-lutidine in DMF at 80 °C gave no dehydrogenation products, although in acetonitrile gave 7% of DMS after 4 h with no increase in yield after 12 h (Table 2, entry 4). Palladium acetate (Pd(OAc)2) in DMF gave DMS only in very low yields at 80 °C, but when the reaction temperature was increased to 120 °C DMS 4 only was formed in approximately 20% yield (Table 2, entries 5 and 6; Scheme 2). With unreacted limonene remaining, this gave a yield of >90% (based on remaining unreacted starting material).
 | ||
| Scheme 2 Reaction conditions for the conversion of limonene 1 to DMS 4 used in further optimisations (Fig. 1). | ||
When the same conditions as for entry 6 (base and temperature) had been used with Pd(OTFA)2 (Table 1, entry 9) an almost identical result was observed. Further experiments using Pd(OAc)2 at 2 mol% and 10 mol% (Table 2, entries 7 and 8) gave less DMS or no products. The reaction was also performed under an oxygen atmosphere without CuCl2 (Table 2, entry 9): 11% of DMS was produced after 9 h, with no further increase in yield after 12 h. The same level of conversion to 4 was reported previously when using 0.5 equiv. CuCl2 under 0.25 MPa of oxygen.12
Reaction monitoring and further optimisations
Using Pd(OAc)2 (5 mol%), CuCl2 (2 equiv.), 2,6-lutidine (3 equiv.) and a reaction temperature of 120 °C the progression of the reaction (Scheme 2) was monitored over 40 h, which indicated that after 4 h the conversion was complete (approx. 20% of DMS only). In addition there was no apparent extended induction period as previously reported at 80 °C, perhaps reflecting the use of higher reaction temperatures.12 The addition of more of the key reagents was investigated (Fig. 1A).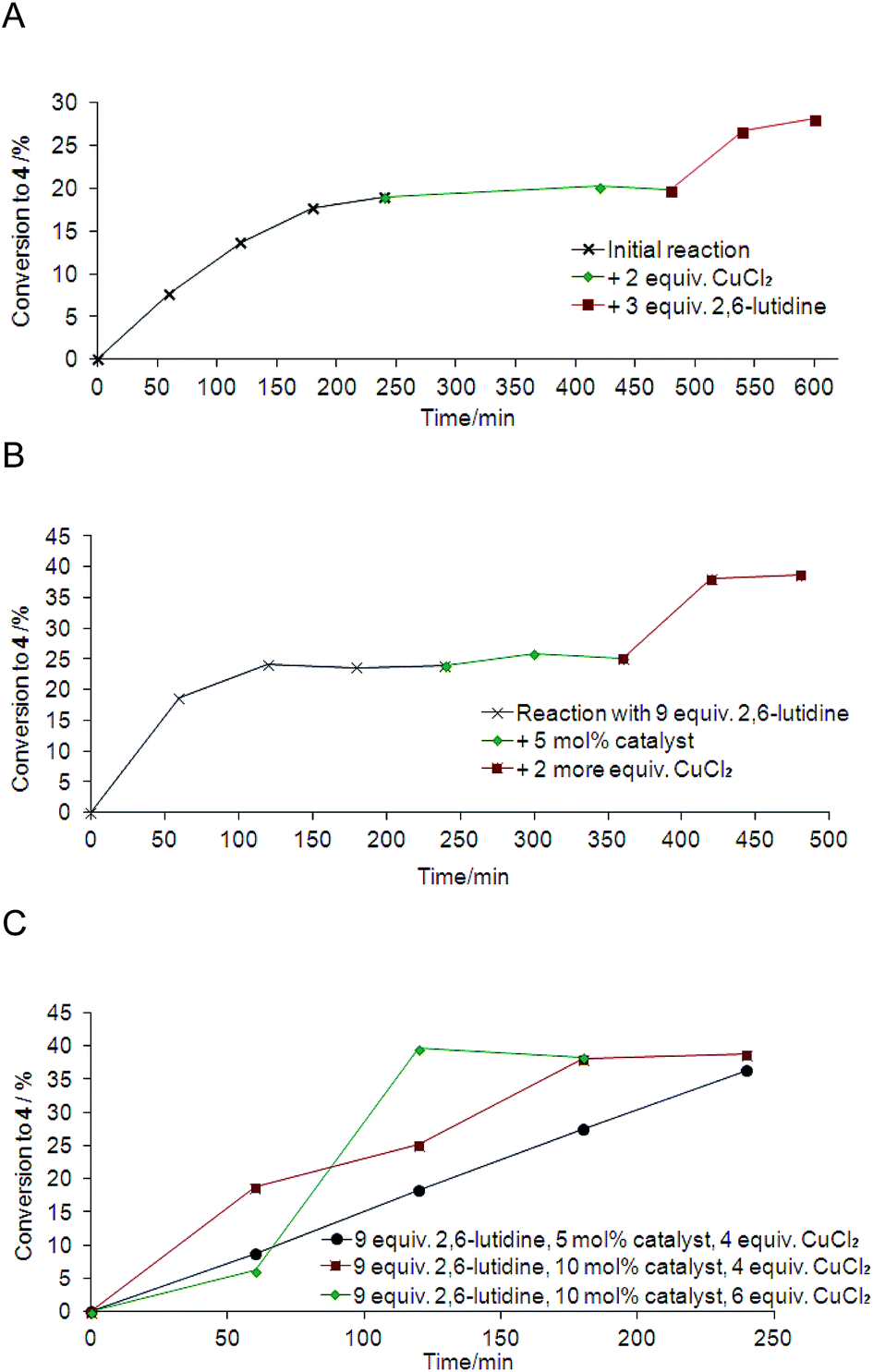 | ||
| Fig. 1 Reaction monitoring. (A) After 4 h the addition of CuCl2 (2 equiv.), and after a further 2 h the addition of 2,6-lutine (3 equiv.); (B) reaction with 9 equiv. 2,6-lutidine: after 4 h the addition of another 5 mol% Pd(OAc)2, and after a further 2 h the addition of more CuCl2 (2 equiv.); (C) reaction profile using 9 equiv. 2,6-lutidine with different amounts of catalyst (5–10 mol%) and oxidant CuCl2 (4–6 equiv.). | ||
Further addition of oxidant CuCl2 (2 equiv.) after 4 h had no effect, but the addition of 2,6-lutidine base (3 equiv.) led to the production of more DMS, increasing the overall yield to 28%. The role of the non-coordinating base such as 2,6-tBu2py and 2,6-lutidine is to remove up to 4 equivalents of HCl generated during the reaction, and was reported previously to enhance the selectivity in the dehydrogenation reaction towards DMS 4. Here it appeared that the removal of remaining HCl, by the addition of >3 equivalents of 2,6-lutidine, could account for the improved conversion to DMS. Therefore, the reaction progression was then followed using initially 2,6-lutidine (9 equiv.) and after 4 h a further 5 mol% of Pd(OAc)2 catalyst was added (no effect was observed), and then after a further 2 h more CuCl2 oxidant (2 equiv.) was added, giving DMS 4 in 38% yield (Fig. 1B). Finally, three further experiments were performed using: 2,6-lutidine (9 equiv.) catalyst Pd(OAc)2 (5 mol% as previously) and oxidant CuCl2 (4 equiv.), and with increased catalyst Pd(OAc)2 (10 mol%) (Table 2, entry 10) and oxidant CuCl2 (6 equiv.). All reactions lead to the formation of DMS in approximately 40% yield, but with less catalyst and oxidant it took 5 h, compared to 2–3 h. The increased amount of 2,6-lutidine in the reaction as well as having a role in the neutralisation of HCl formed, may also help to stabilise the active catalytic species and prolong its lifetime. In all cases the selectivity of the reaction was exceptional, leading only to the formation of DMS 4 (with limonene starting material remaining).
Comparing the reaction profile data (Fig. 1) to that in Table 2, where an increase in palladium catalyst Pd(OAc)2 inhibited the reaction (entry 10), an increase in the amount of catalyst combined with more oxidant CuCl2 to oxidise the catalyst, clearly had a beneficial effect. This highlights that the complexity of the reaction, which involves up to 12 steps, makes the outcome of changing one reagent very difficult to predict. Nevertheless, reaction conditions have been identified to give a method for the conversion of limonene selectively to DMS 4 in 40% yield. The generation of a single dehydrogenated product simplifies significantly the purification of the reaction. Indeed, since 3 and 5 have boiling points within 1–4 °C of DMS 4, the production of a single dehydrogenation product is particularly valuable. The dehydrogenation reaction was scaled-up (to 3 ml D-limonene) using the reaction conditions in Table 2, entry 10, (Scheme 2, Fig. 1C) and the product DMS 4 readily separated by vacuum distillation in 37% isolated yield and >99% purity (no impurities detected). This method of generating DMS could be used in a recirculatory flow reactor, which would recycle the unconverted limonene.
Conclusions
In summary, improved conditions have been developed for the selective dehydrogenation of limonene to the monomer DMS 4 in a highly selective procedure. By performing the reaction at 120 °C with Pd(OAc)2 catalyst, 2,6-lutidine as a non-coordinating base, and CuCl2 as an oxidant approx. 40% of DMS was formed. Notably, no products resulting from the isomerization of the exocyclic double bond were observed under these conditions.Experimental
General experimental details
Reagents were used as supplied: D-limonene (Aldrich, 97%); palladium(II) trifluroroacetate; palladium(II) acetate (Aldrich, 98%); palladium(II) chloride (Alfa Aesar, 47% of Pd); anhydrous copper(II) chloride (Alfa Aesar, 98%); 2,6-di-tert-butylpyridine (Aldrich, >97%); N,N-diisopropylethylamine (Aldrich, >99%); 2,2,6,6-tetramethylpiperidine (Aldrich, >99%); 2,4,6-collidine (Aldrich, 99%), except 2,6-lutidine (Aldrich, >99%) which was dried with 4 Å molecular sieves for 18 h prior to use. Solvents were used as supplied: DMF (Acros, 99.8%); DMSO (Aldrich, 99.6%), and all other solvents (Aldrich, 99.9%).Dehydrogenation of D-limonene 1 to DMS 4
The following method was used, with the modifications outlined above. D-limonene (1 equiv.) was added to a stirred solution of PdX2 (catalyst, 5 mol%), CuCl2 (2 equiv.) and base (3 equiv.), unless indicated otherwise, in anhydrous solvent. The reaction was heated for the time and at the temperature indicated. The reaction was followed by GC analysis (as previously described12) using a Hewlett Packard HP4890-A GC with a high performance capillary column HP-5 (crosslinked 5% PH ME siloxane) (30 m × 0.32 mm × 0.25 μm), with He carrier gas and a flow rate of 2 ml min−1. The injector was operated at 250 °C in split mode (ratio 1:20). The oven was heated to 50 °C and then increased at 1 °C min−1 to 80 °C. Calibrations were performed for 1–6, with retention times of approximately 1 16.9 min, 2 14.8 min, 3 22.7 min, 4 24.0 min, 5 20.3 min and 6 14.2 min.
Scaled up reaction
D-limonene (3.00 ml, 18.5 mmol) was added to a mixture of PdOAc2 (10 mol%), CuCl2 (4 equiv.), 2,6-lutidine (9 equiv.) and anhydrous DMF (45 ml). The reaction mixture was heated at 120 °C for 3 h, and DMS 4 isolated by vacuum distillation (30 mmHg, 80 °C) in >99% purity (0.904 g, 37%).Acknowledgements
The authors are grateful to the National Council of Science and Technology of Mexico (CONACYT) for providing a stipend for S. A. S.-V.Notes and references
- S. A. Sanchez-Vazquez, H. C. Hailes and J. R. G. Evans, Polym. Rev., 2013, 53, 627 CrossRef CAS.
- R. J. Grau, P. D. Zgolicz, C. Gutierrez and H. A. Taher, J. Mol. Catal. A: Chem., 1999, 148, 203 CrossRef CAS.
- C. Zhao, W. Gan, X. Fan, Z. Cai, P. J. Dyson and Y. Kou, J. Catal., 2008, 254, 244 CrossRef CAS PubMed.
- P. A. Weyrich and W. F. Holderich, Appl. Catal., A, 1997, 158, 145 CrossRef CAS.
- R. Neumann and M. Lissel, J. Org. Chem., 1989, 54, 4607 CrossRef CAS.
- M. A. Martín-Luengo, M. Yates, M. J. Martínez Domingo, B. Casal, M. Iglesias, M. Esteban and E. Ruiz-Hitzky, Appl. Catal., B, 2008, 81, 218 CrossRef PubMed.
- C. Fernandes, C. Catrinescu, P. Castilho, P. A. Russo, M. R. Carrott and C. Breen, Appl. Catal., A, 2007, 318, 108 CrossRef CAS PubMed.
- C. Breen and A. J. Moronta, Clay Miner., 2001, 36, 467 CrossRef CAS PubMed.
- A. K. Mukhopadhyay, Industrial Chemical Cresols and Downstream Derivatives, Marcel Dekker, New York, 2005 Search PubMed.
- R. B. Seymour, F. F. Harris and I. Branum, Ind. Eng. Chem., 1949, 41, 1509 CrossRef CAS.
- R. W. Lenz, J. E. Sutherland and L. C. Westfelt, Makromol. Chem., 1976, 177, 653 CrossRef CAS.
- P. Horrillo-Martínez, M. A. Virolleaud and C. Jaekel, ChemCatChem, 2010, 2, 175 CrossRef.
- B. M. Trost and P. J. Metzner, J. Am. Chem. Soc., 1980, 102, 3572 CrossRef CAS.
- J. E. Bercaw, N. Hazari and J. A. Labinger, J. Org. Chem., 2008, 73, 8654 CrossRef CAS PubMed.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: GC calibration data and example GC analysis traces. See DOI: 10.1039/c4ra11558a |
| This journal is © The Royal Society of Chemistry 2014 |