
Aniline tetramer embedded polyurethane/siloxane membranes and their corresponding nanosilver composites as intelligent wound dressing materials†
Reza Gharibia,
Hamid Yeganeh*a,
Hoshyar Gholamia and
Zuhair M. Hassanb
aIran Polymer and Petrochemical Institute, P.O. Box: 14965/115, Tehran, Iran. E-mail: h.yeganeh@ippi.ac.ir; Fax: +98 2144787023; Tel: +98 2148662447
bDepartment of Immunology, School of Medical Science, Tarbiat Modares University, P.O. Box: 14115-331, Tehran, Iran. E-mail: hasan_zm@modares.ac.ir
First published on 13th November 2014
Abstract
The preparation of functional intelligent wound dressing membranes with features including: excellent dimensional stability in both dry and hydrated states, preserving a moist environment over the wounded area by self-adjustment of their permeability in response to wound moisture level, a broad spectrum of antimicrobial activity towards different microorganisms, promising antioxidant (radical scavenging) efficiency and significant activity for stimulation of fibroblast cells growth and proliferation is reported in this work. For the preparation of dressing membranes, NCO-terminated polyurethane prepolymers and a CHO-terminated aniline tetramer are functionalized with silane groups. Polyurethane/siloxane networks are prepared through a sol–gel reaction of these components at different weight ratios. The corresponding nanosilver composites are simply prepared by a redox reaction through impregnating the membranes with a silver salt solution. Based on various in vitro tests, it is revealed that some of the prepared membranes have the basic characteristics of ideal dressings, while they can actively alleviate the healing of tissue in the wounded area.
Introduction
A wound can be described as a defect or a rupture in the skin, resulting from physical or thermal damage or due to the presence of an underlying medical or physiological condition.1 When skin is damaged, a wound dressing is used to serve as a physical barrier against external forces, dust and bacteria. Also, the dressing should manage (retain or remove) the exudates in the wounded area, since the maintenance of a moist and warm wound bed is widely accepted as the best environment for effective wound healing.2 Non-toxicity, biocompability, oxygen permeability, good handling, proper adhesion and sufficient resilience and hardness are additional characteristics of ideal wound dressing materials.1 Preparation of wound care dressings having the maximum number of aforementioned features is a continuing challenge.Accelerating the wound healing processes is an important issue that has attracted much attention in recent years. It is anticipated that modern wound dressings should change from traditional materials to active and functional materials. It is expected that these dressings not only protect wounded area, but also interact with the wound bed appropriately to encourage the healing process.3 Stimulation of proliferation and migration of keratinocytes by transdermal delivery of growth factors like epidermal growth factor (EGF) is an example of accelerated wound healing process.4 Indeed, the wound healing process is complex and involves the interactions of many different types of cells and matrix components to establish a provisional tissue and eventually a complete regenerated epidermis. Therefore, application of growth factors may suffer from some limitations such as high manufacturing costs, low bioavailability and imperfect effect of a single growth factor on wound healing processes. As well, inactivation of growth factors at the chronic wound bed due to proteolytic degradation by protease activity is another concern in this method.3
It seems that constructing wound dressing materials that can help body to heal itself is the preferred choice in this emerging subject. To full fill this goal; three simultaneous strategies are followed in the present study. The first concern is preventing the effect of free radicals. Free radicals are present to a great extent in all types of trauma.5,6 The process of wound healing can be hampered by the presence of free radicals; therefore, quenching radical activity can prevent further damage to wound surrounding cells and consequently increase the rate of healing process.7 Some of recent studies have shown promising performance of antioxidant and radical scavenging properties of conducting polymers such as polyaniline and oligoaniline derivatives.8,9 Utilization of these unique aspects of oligoaniline is examined in the present work.
The second concern is reducing the bacteria influence on damaged tissue. It is confirmed that reducing infection and subsequently decreasing the wound exudates by application of effective antimicrobial agents on infected wounds can accelerate the healing process.10
In recent studies it is emerged that polyaniline and its derivatives may exhibit broad spectrum of biocidal activity.11 Gizdavic-Nikolaidis and co-workers12 have shown that in addition to polyaniline and its functionalized analogous, the low molecular weight oligomeric types of these materials have antibacterial activity as well. Therefore, utilization of this inherent property of oligomeric aniline moieties (aniline tetramer) is considered in the present work. The common practice for utilization of dressing materials with antibacterial activity is loading of different antimicrobial agents such as iodine,13 quaternary ammonium salts,14,15 zinc oxide16 and silver17 in wound dressings. Due to a broad spectrum of antimicrobial activity and low bacterial resistance, application of silver nanoparticles has currently attracted considerable attention. To broaden and increase the efficiency of biocidal activity of dressing membranes, the incorporation and utilization of silver nanoparticles has also been considered in the present study. Two approaches are commonly employed for the preparation of polymer–silver nanocomposites. In the first method, as-prepared silver nanoparticles can be mixed with polymeric matrix. Agglomeration and improper distribution of silver nanoparticles are the main drawbacks of this approach.18 In the second approach, the polymeric matrix is swollen with the solution of silver salt. In situ reduction of silver ions by a reducing agent like NaBH4 leads to homogeneous dispersion of silver nanoparticles into the polymeric matrix. Utilization of reducing power of embedded oiligoaniline19,20 into the polymeric matrix is a fascinating method that has been used in the present work for rapid and efficient production and distribution of silver nanoparticles into polymeric matrix of the dressing.
Electrical stimulation has been found to be an effective technique to promote wound healing process.21 Recent studies have shown that the electrical stimulation may increase protein, collagen and DNA synthesis. Cellular activities involving cell adhesion, migration, proliferation and differentiation can be also regulated by this type of stimulation.22–24 To have these advantages, many researchers have tried to incorporate different kinds of conducting polymers into biomaterials, mainly for tissue engineering application. In this regard, use of oligoaniline with accept' electrical conductivity and good processability has attracted extensive interest.25–27 It is worth mentioning that these advantages can be attained even in the absence of an external electrical stimulation.28–30 Accelerated proliferation, adhesion and growth of different cells such as osteoblasts and preosteoblastic MC3T3-E1 cells on polymeric materials containing electroconductive oligoaniline were reported by Yen Wei and co-workers.31–33 Neurite extension and nerve repair accelerating without any need of external electrical signals have also been reported for matrix-containing electroactive polymers.23 Primary dermal fibroblast and keratinocytes are other important cells, especially for skin wounds, have been examined for their accelerated growth on electroactive matrix by Ateh and Shin groups.34–36 Therefore, utilization of this possible advantage is another goal of this study.
Synthesis, characterization and assessment of hybrid organic–inorganic polyurethane networks embedded with aniline tetramer and corresponding nanocomposites containing silver nanoparticles are described in the present study. Based on different in vitro methods it is tried to show that regulation of wound exudates, facilitation of cell proliferation, combating bacterial contamination and having radical scavenging ability are versatile means to boost wound healing process.
Experimental
Materials
Poly(ethylene glycol) (PEG, Mn 2000) from Aldrich was dried by azeotropic distillation with dry toluene just before use. Tetraethoxysilane (TEOS), (3-aminopropyl)trimethoxysilane (APS), N-phenyl-1,4-phenylenediamine, ammonium persulfate, glutaraldehyde (GA) (50% by wt in H2O) and camphorsulfonic acid (CSA) were purchased from Aldrich. Isophorone diisocyanate (IPDI) from Merck was purified via vacuum distillation. N,N-Dimethylformamide (DMF) and toluene were distilled over CaH2 and sodium, respectively. All other chemicals were of analytical grade and used as received. S. aureus (ATCC 6538), E. coli (ATCC 25922) and P. aeruginosa (ATCC 15449) bacteria and C. albicans (ATCC 10231) were purchased from Iranian Research Organization for Science and Technology (IROST). Mouse L929 fibroblast cells were received from Pasteur Institute of Iran and used as obtained.Synthesis of silane-terminated polyurethane-prepolymer (Si-PU)
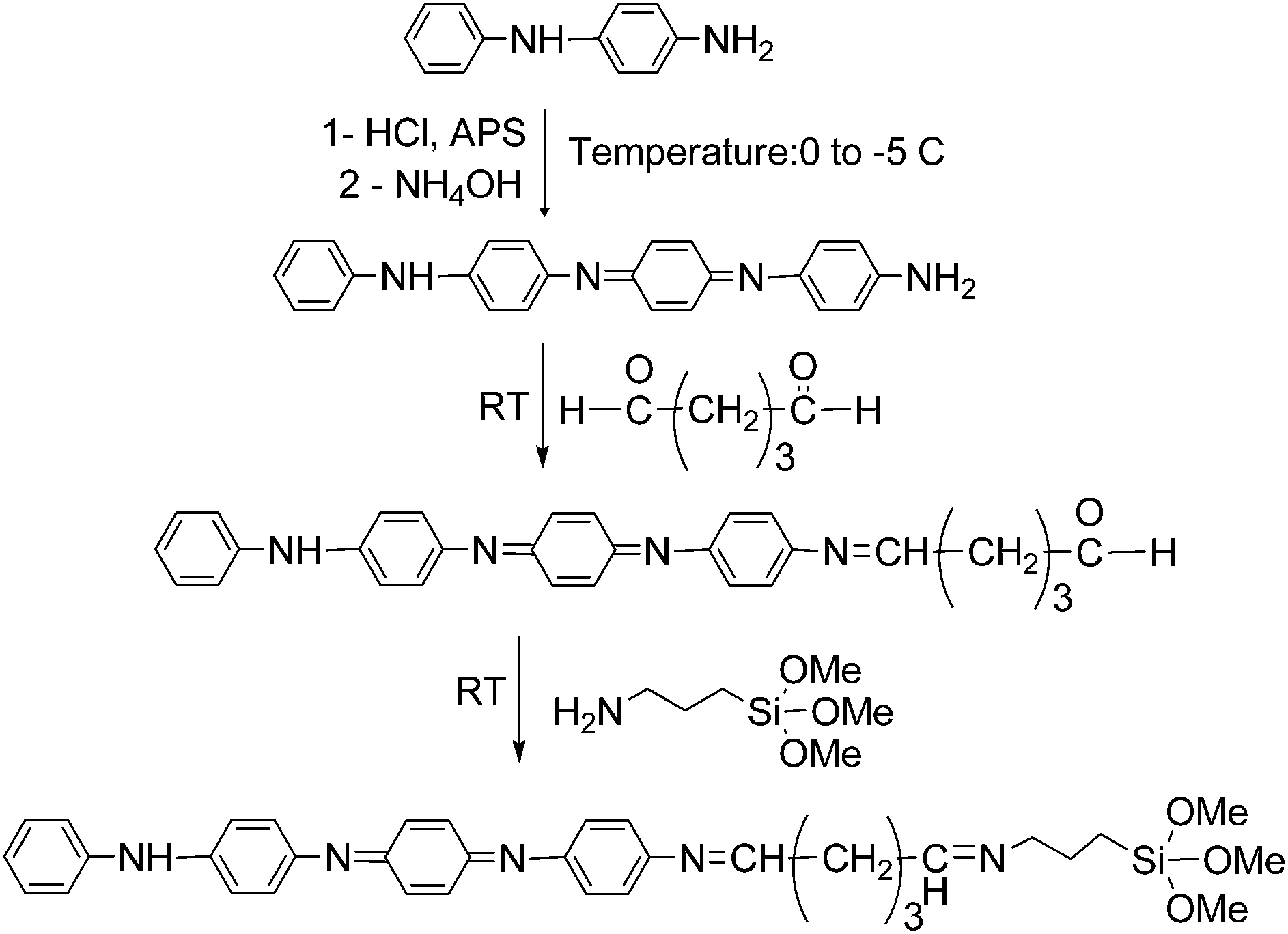
A three-necked polymerization reactor equipped with a mechanical stirrer, condenser, dropping funnel and a nitrogen inlet was charged with PEG (25.00 g) and dried DMF (25 ml). A solution of IPDI (5.56 g) in DMF (15 ml) was slowly dropped into the reactor during 30 min under ambient temperature. While stirring, the temperature was increased to 85 °C. The reaction was continued until the free NCO content of the product reached the calculated theoretical value. The heating mantel was removed, and the reactor content was cooled to room temperature. Then, APS (4.48 g) was dropped into the reactor and the mixture was stirred at room temperature for 30 min, and then the temperature increased to 50 °C for 3 h. The reaction product with a pre-determined solid content was transferred to a glass bottle, sealed and kept in refrigerator.Synthesis of aniline tetramer (AT) and glutaraldehyde functionalized aniline tetramer (GA-AT)
AT was prepared via oxidative coupling of N-phenyl-1,4-phenylenediamine by ammonium persulfate.37 It was purified by washing with 1 mol L−1 HCl and plenty of distilled water. After depoping with 1 mol L−1 NH4OH the filtered AT was washed again several times with distilled water until the filtrate was neutral (checked by pH paper). Then, it was further purified with acetone in a Soxhlet Extractor. The completely vacuum dried sample was used for the preparation of GA-AT. This compound was also prepared according to the procedure described in the literature37 and purified by several washing with cold diethyl ether and vacuum dried.Synthesis of silane-terminated aniline tetramer (Si-AT)
GA-AT (0.50 g) was dissolved in DMF (5 ml) and then APS (0.20 g) was added into the solution. The reaction mixture was stirred at room temperature for 2 h. The prepared product was refrigerated for further use.Preparation of wound dressing membranes (EAPU1-3)
The mixture of Si-PU, TEOS, various amounts of Si-AT and an appropriate amount of water was dissolved in DMF according to the formulations given in Table 1. The mixture was stirred vigorously for 1 min, evacuated to remove any trapped air and then cast slowly into a clean Teflon mold. The mold was kept at room temperature overnight. It was then heated in an oven at 70 °C for 10 h and at 100 °C for 2 h. The resulting membrane was finally subjected to high vacuum at 70 °C to ensure removal of any residual solvent. The thickness of the membrane was adjusted by controlling the solid content of mixture. The cured membrane was cut to the desired shape for further experiments. Some of the prepared membranes were doped by immersion in a CSA (2 mol l−1) solution for 24 h.| Sample | Si-PU (g) | Si-AT (g) | TEOS (g) | H2O (g) | Gel Content (%) |
|---|---|---|---|---|---|
| a According to analysis of variances P-values of <0.05 were considered significant. The difference between quantities with similar superscripts is not significant (p ≥ 0.05) for data of each column. | |||||
| NEPU | 1.00 | — | 0.20 | 0.15 | 97.7 ± 0.7a |
| EAPU1 | 1.00 | 0.18 | 0.20 | 0.15 | 97.8 ± 0.4a |
| EAPU2 | 1.00 | 0.30 | 0.20 | 0.15 | 97.6 ± 0.1a |
| EAPU3 | 1.00 | 0.42 | 0.20 | 0.15 | 97.6 ± 0.6a |
For comparison, a none-electroactive polyurethane membrane (NEPU) was also prepared through the similar procedure using all ingredients except Si-AT component.
It is worth to mention that the final dressing membranes were subjected to the solvent extraction/purification procedure by immersion into ethanol (70% w/w) solvent for 24 h and subsequently into distilled water for another 24 h before further characterization and assays.
Preparation of nanosilver containing wound dressing membranes (EAPU1-3-Ag)
NEPU and CSA doped EAPU membranes (1 × 1 cm) were immersed into a beaker containing silver nitrate solution (0.4 g l−1) for 24 h. Then, it was removed and washed with plenty of distilled water. Silver nanoparticles (Ag NPs) loaded film was dried at ambient temperature under vacuum and used for subsequent studies.Equilibrium water absorption (EWA) and equilibrium water content (EWC) of membranes
The completely dried sample was accurately weighed and soaked in PBS at room temperature until the equilibrium swelling was attained (about 48 h). The weight of swelled membrane was determined after being gently wiped with filter paper to remove the surface liquid. EWA% and EWC% were determined using the following equations: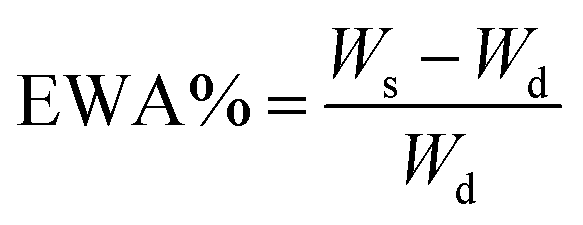
where, Wd and Ws designated the weights of dry and swelled membrane, respectively.
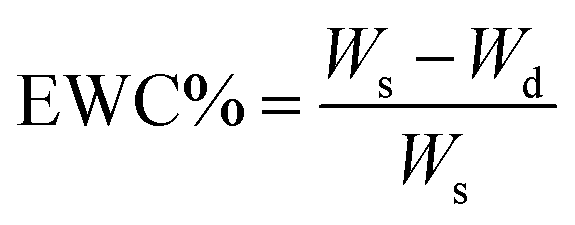
Fluid handling capacity of membranes
The fluid handling capacity (FHC) of the dressing membranes at certain surface area and thickness was calculated as the sum of the water vapour transmission rate (WVTR) and absorbance capacity (AC) determined via a technique described in BS EN13726-1, 2002.To mimic the typical environment of serum and wound exudate, a solution of sodium/calcium chloride containing 142 mmol l−1 of sodium ions and 2.5 mmol l−1 of calcium ions was used.
For measuring the AC of samples a completely dried membrane with thickness of (0.1 mm) was soaked into a beaker containing the test solution. The weight of swelled membrane was determined after being gently wiped with filter paper to remove the surface liquid. AC was calculated using the following equation:
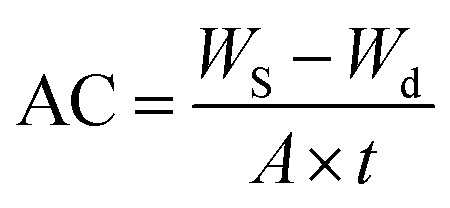
WVTR of membranes was determined under two different states of conditions. In the first state the membrane was not in direct contact with liquid and it was just exposed to moisture vapour. In the second state the membrane was placed in direct contact with the test solution.
Five samples of each dressing of known weight were applied on the cup mouth containing test fluid. The cups were weighed and placed in an incubator at 37 ± 2 °C for a period of 24 h. At the end of the test, the cups were removed from the incubator, allowed to equilibrate to room temperature and reweighed. The loss in weight due to the passage of moisture vapour through the dressing was determined using the following equation:

In vitro cytocompatibility assays
Cytocompatibility of the prepared dressings were evaluated by microscopic study of L929 fibroblast cells morphology after direct contact with samples as well as tetrazolium dye-based colorimetric assay (MTT assay) according to our previously reported procedure.14The samples were sterilized by incubation at 120 °C for 15 min before each test. In “direct contact test”, the evaluation of cells morphology present on the interface of samples and culture plate was performed after incubation time of 48 h.
To check whether the cytotoxic agents may possibly leach out from the wound dressing membranes, the freshly synthesized samples were immersed in the culture medium for 3 days at 37 °C and then MTT assay was performed on extracted leachates using L929 fibroblast cells.
The percentage of relative cell viability was calculated according to following equation:

Evaluation of cell proliferation on dressing membranes
The number of cells attached onto the surface of dressing membranes, with and without electroconductive AT moieties, was evaluated in order to estimate the possible participation of dressing material on cell growth and proliferation. For this purpose, cell attachment and proliferation on the prepared membranes were quantitatively assessed using MTT assay. At first, the sterilized membranes were cut in a size to completely cover the bottom of each well of a 96-well culture plate. Then, the culture medium containing 5 × 103 L929 fibroblast cells were placed on the membrane and incubated at 37 °C for 45 min to enable the cells to attach onto the membrane surface. Subsequently, the membrane per cell constructs were provided with more culture medium for 1, 3, and 5 days, respectively. The seeded membranes were then washed with PBS to remove unattached cells and were transferred into new culture plates. Cells were harvested after 3 min incubation with 0.25% trypsin and 1 mM EDTA at 37 °C and the quantification was made by the MTT assay according to the procedure implemented for cytocompatibility assessment.The spatial distribution and morphology of the L929 cells attached onto the surface of the membranes were assessed after staining of cells by a fluorescent dye. For this purpose, the above mentioned procedure was followed. After seeding of cells on the samples surface and their incubation for specifically determined times of 1, 3, and 5 days, the samples were washed three times with PBS. The adhered cells were fixed with 2.5% glutaraldehyde at room temperature for 8 min. The cells were dyed with DMSO containing 2% of curcumin for 8 min; followed by washing the samples with PBS several times. The stained cells were observed by an inverted microscope (TE2000-U, Nikon).
Antimicrobial activity of membranes with and without Ag NPs
Antibacterial activity of the prepared membranes was studied using “colony forming count” method. Bacteria at inoculated concentration of 2 × 108 CFU ml−1, and membranes with the dimensions of 1 cm × 1 cm were used throughout the tests. For those samples containing chemically bonded AT moieties the procedure reported in ASTM E 2180-07 was followed. 18 h cultures prepared from three microorganisms including S. aureus (ATCC 6538), E. coli (ATCC 25922) and P. aeruginosa (ATCC 15449) and C. albicans (ATCC 10231) in tryptic soy broth. Microbial broth cultures were adjusted to 3 × 108 cells per ml with a spectrophotometer. 1 ml of these cultures was inoculated into Erlenmeyer flasks containing 100 ml molten agar slurry (0.3 g agar–agar, 0.85 g NaCl, 100 ml water). A thin layer of the inoculated agar slurry (100 μl) is pipetted onto the test and untreated control materials (triplicate samples). After the specified contact time (24 h commonly used), surviving microorganisms are recovered via elution of the agar slurry inoculum from the test substrate into neutralizing broth and extracted via vortexing that provide complete removal of the inoculum from the test polymers. Serial dilutions were made, and then spread plates were made of each dilution. Agar plates and dilution broths are incubated for 48 ± 2 h at 37 °C as optimal temperature for test organisms. Microbial colonies from each dilution series were counted and recorded. The percentage of microorganism reduction was calculated according to the following equation: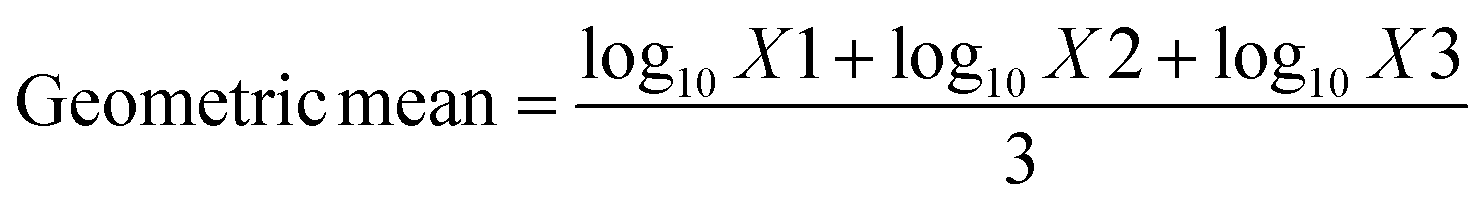
where x designates number of organisms recovered from the incubation period control or incubation period treated samples, a designates the antilog of the geometric mean of organisms recovered from the incubation period control samples and b designates the antilog of the geometric mean of organisms recovered from the incubation period treated samples.

For those samples containing Ag NPs, the “shaking flask” method was followed. The membranes (triplicate samples) were seeded with 10 ml determined colony forming units per ml (CFU ml−1) of either E. coli, S. aureus, P.aeruginosa and C. albicans. The solutions containing the samples were incubated in a shaking incubator at 37 °C for 24 h. For correct counting of the grown bacterial colonies, 0.1 ml of the treated solution was diluted with double distilled water to a suitable volume and then, the diluted solution was spread on a nutrient agar plate and incubated at 37 °C for 12 h. A NEPU sample (blank sample) was also studied as positive control. The counts were used to calculate the surviving number of bacteria. The percentage of bacterial reduction was calculated using the following equation:

Antioxidant efficiency of membranes
The antioxidant efficiency of specimens were tested by measuring their capacity to scavenge 1,1-diphenyl-2-picrylhydrazyl (DPPH) as a stable free radical, using the method reported by Serpen with minor modification.38 The samples in doped state were powdered by crushing in liquid nitrogen and then dispersed in methanol. The sample (1 mg) and DPPH (3.0 ml, 100 μM) were dispersed in methanol and incubated in a dark place for 15 min. The wavelength scanning was performed on the samples using a UV-vis spectrophotometer. The DPPH degradation was calculated using the following equation:where AB being the absorption of the blank (DPPH + methanol) and AS the absorption of the sample (DPPH + methanol + sample).
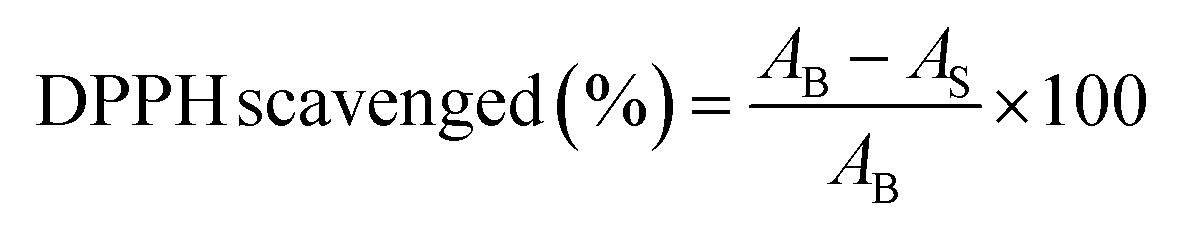
Measurements
NCO content of polyurethane prepolymer was determined according to the procedure reported in ASTMD-2572. FTIR spectra were obtained using a Bruker IFS 48 instrument. All spectra were obtained in air as a function of time with 16 scans at a resolution of 4 cm−1 and a spectral range of 500–4000 cm−1. The UV-visible spectra were monitored on a Bel photonics UV-vis spectrophotometer (LGS 53).For the measurement of the electrical conductivity, the membranes, at both dry and hydrated states, were used in circular shape with thickness of about 1 mm. The conductivity was measured using a home-made four-point probe instrument utilizing following equation:

Dynamic mechanical thermal analysis (DMA) was carried out with a Tritec 2000 DMA instrument in the temperature range −100 to 150 °C at a heating rate of 3 °C min−1 and a frequency of 1 Hz. The dimension of each sample was 30 × 10 × 1 mm3. The values of storage modulus, loss modulus and tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ versus temperature were recorded for each sample.
δ versus temperature were recorded for each sample.
The measurement of tensile strength, elongation-at-break, and modulus of samples under dry and hydrated states were carried out using a tensile tester (Instron 6025) with a crosshead speed 50 mm min−1. Samples were cut into bars of each 50 mm length and 5 mm width. The test was performed at room temperature. For each sample five specimens were tested.
The gel content of cured membranes was evaluated. For this purpose the membranes were dried under vacuum for 24 h at room temperature and weighed. Then, the samples were extracted by THF in a Soxhlet extractor for 24 h. The insoluble part was dried at 50 °C and weighed. The gel content was defined as follows:

The size of Ag NPs was evaluated by atomic force microscopy (AFM) in contact mode. For this purpose, the EAPU membrane containing Ag NPs was placed in distilled water for 72 h. A few drops of the extracted solution were placed on a mica slide. The residue, which was obtained after slow evaporation of solvent, was examined by AFM. The silver atom content and its distribution map were evaluated by a Tescan scanning electron microscope (Vega II, Czech) equipped with an energy dispersive X-ray analyzer system (EDX, Oxford Instrument, INCA, England).
Surface hydrophilicity of membranes was determined by measuring the water droplet contact angle at six different positions of membrane surface for each sample. The contact angle was determined by running Image J 1.44p software on digital pictures taken from interfaces of membranes and droplets. The values reported are an average of six measurements.
Statistical analysis
Statistical analyses were performed via PASW Statistics program package, version 18 (SPSS, Chicago, IL). Comparison of obtained data for different samples was performed with one-way analysis of variance with Tukey or Bonferroni post hoc tests. The significance level was set at p > 0.05.Results and discussions
Incorporation of electroactive aniline tetramer moieties into the backbone of polyurethane membranes has been considered in this work. It is worth mentioning that polyurethane backbone has been intentionally selected in the present work since this class of polymers has been widely applied as wound dressing materials.39 The most polyurethane-based dressings currently available in the market are in the form of dense membranes, which may exhibit certain weaknesses including low moisture vapour/gas transmission rate and poor exudate absorption capability. For clinical cases where exuding wounds are encountered, these drawbacks have been found to cause exudate accumulation that disturbs granulation, risks of infectious complications, and thus intensifies nursing care. To overcome this disadvantage and improve healing of wounded area a polyurethane with high capacity for absorbing water through hydrogen bonding was synthesized.40 A distinct disadvantage for such polyurethane hydrogel wound dressings would be poor mechanical properties, especially at hydrated state.15 Improving the mechanical strength through crosslinking reaction was considered as a solution to combat this problem. One attractive strategy for enhancing the mechanical properties of polyurethane matrices is introduction of inorganic siloxane moieties into the backbone of polyurethane through sol–gel reaction of chemically embedded alkoxysilane functions.41,42 This strategy was also adopted in the present work.Synthesis and spectroscopic characterization of dressing membranes
Schemes 1, 2 and S1† show our preferred synthetic route for the preparation of functional dressing membranes with balanced mechanical property and high exudates absorption ability. The reaction of one mol of PEG with 2 mol of IPDI under the appropriate condition led to the formation of the NCO-terminated polyurethane prepolymer (Scheme 1). Then, APS (2 mol) was added and the reaction continued until no free NCO peak at 2230 cm−1 was detected in the FTIR spectrum of sample taken from reaction mixture (Fig. S1†). | ||
| Scheme 1 Synthesis route for the preparation of Si-PU. | ||
 | ||
| Scheme 2 Synthetic route for the preparation of AT and Si-AT. | ||
AT was synthesized via oxidative coupling of N-phenyl-1,4-phenylenediamine in the presence of ammonium persulfate. To incorporate AT segments into the final networks silane-functional aniline tetramer was prepared as shown in Scheme 2. GA was used as the coupling agent between AT and APS. FTIR spectra of AT, GA-AT and Si-AT were shown in Fig. S2a–c.†
To confirm that the functionalization reactions performed on AT would not affect its electroactivity, the UV-visible spectra of AT, GA-AT and Si-AT were recorded in DMF (Fig. S3†).
The wound dressing membranes with different concentrations of electroactive AT counterpart were prepared through mixing of ingredients as tabulated in Table 1. The reaction of alkoxysilane moieties and formation of siloxane network (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O–Si) through hydrolysis and condensation reaction was triggered by addition of water under acidic condition. The probable structure of final network is shown in Scheme S1.† To control the crosslink density and tune the mechanical property of final networks through formation of siloxane domain, a constant weight of TEOS was added to all formulations. The high value for gel content of the prepared networks (Table 1) showed successful progress of sol–gel reaction. The presence of Si–O–Si peak at about 1050 cm−1 in the ATR-FTIR spectra of the cured membranes (Fig. S4a and b†) was also a confirmation in the formation of desired networks.
Si–O–Si) through hydrolysis and condensation reaction was triggered by addition of water under acidic condition. The probable structure of final network is shown in Scheme S1.† To control the crosslink density and tune the mechanical property of final networks through formation of siloxane domain, a constant weight of TEOS was added to all formulations. The high value for gel content of the prepared networks (Table 1) showed successful progress of sol–gel reaction. The presence of Si–O–Si peak at about 1050 cm−1 in the ATR-FTIR spectra of the cured membranes (Fig. S4a and b†) was also a confirmation in the formation of desired networks.
Viscoelastic and tensile properties of wound dressing membranes
Viscoelastic properties and different types of transitions and relaxations related to the structure of polyurethane/siloxane membranes with various amounts of Si-AT were analyzed by DMA method. The tests were performed in tensile mode as a function of temperature from −100 to 150 °C. Typically, variations in storage modulus (E′), loss modulus (E′′) and loss tangent (tanδ) as a function of temperature were monitored and three phases including the glassy state, the transition region and the rubbery plateau, were covered for each sample. The results are shown in Fig. 1. All membranes showed two phase structures, since two main transitions were detected in DMA curves. The transition at lower temperature in the range of −39 to −27 °C (from tanδ maximum) was attributed to soft PEG segment. Slightly higher Tg value for PEG segments in the prepared network in comparison to the neat PEG, of the same molecular weight, (−62 °C) showed partial phase mixing in the samples. The transition at higher temperature in the range of 0 to 3 °C was attributed to hard segment domain composed of siloxane domain. Relatively low Tg of hard segment in these samples is most likely due to the low molecular weight of hard domain as compared with similar crosslinked polyurethanes prepared through UV or moisture curing reactions.42–44 The Tg of soft segment was also shifted to higher temperature upon introduction of Si-AT counterpart. This peak was almost merged with hard segment glass transition in sample EAPU3 with the highest concentration of Si-AT. This phenomenon may be related to higher dipole–dipole interaction of ether bonds of PEG domain with aromatic rings of AT. Higher inter- and intramolecular interactions (intermolecular π–π stacking of aniline tetramers' moieties) resulted in an increase in the modulus of rubbery plateau region for EAPU3 in comparison to NEPU. The height of tanδ peak was also decreased and width at half peak height increased in EAPU3. Meanwhile, the decrease in storage modulus at glass transition region was much steeper for NEPU sample than those with Si-AT. These phenomena again confirm the higher phase mixing of networks upon introduction of proper concentration of Si-AT moieties. Increasing the rubbery plateau modulus also showed reinforcement of membrane upon introduction of aromatic planar aniline tetramer rings. Monitoring the variation of storage modulus vs. temperature showed extension of the rubbery plateau region up to 150 °C for all samples. Therefore, the prepared membranes can withstand the high temperature (105 °C) needed for autoclave sterilization procedure.
 | ||
| Fig. 1 DMA curves of the wound dressing membranes. | ||
The dressing materials applied to the wound bed should preserve their dimensional stability during the healing period. To acquire an insight into this important factor, tensile strength of the membranes was assessed at both dry and wet conditions. It is well known that plasticization effect of the absorbed water molecules can dramatically reduce the intermolecular interaction of polar polymeric chains and consequently reduce the overall mechanical strength. Therefore, the tensile test was also performed on fully hydrated samples to confirm the extended applicability of these membranes for the protection of high exuding wounds. The tensile strength, initial modulus, and elongation-at-break of samples were extracted from their stress–strain curves (Fig. 2a and b) and corresponding data are summarized in Table S1.† All the prepared membranes showed general behavior of lightly crosslinked elastomers with suitable mechanical property for wound dressing application. The reinforcing effect of polysiloxane domain produced through hydrolysis and condensation of methoxysilane moieties is responsible for such observed behavior. The tensile strength and initial modulus were increased with increasing the concentration of Si-AT in the formulation of membranes, whereas an opposite trend was detected for the elongation-at-break values. This phenomenon was in accordance with DMA data and it was related to the presence of Si-AT moieties. As it was stated by Wei,45 the aromatic rings of aniline oligomers can stack over each other through strong π–π interactions and consequently increase the strength of the network. It is interesting to note that the reinforcing effect of oligoaniline moieties is more evident in tensile properties at large deformation. In fact, alignment and extension of polyetherurethane chains in rubbery state contribute to the improved performance of AT moieties.
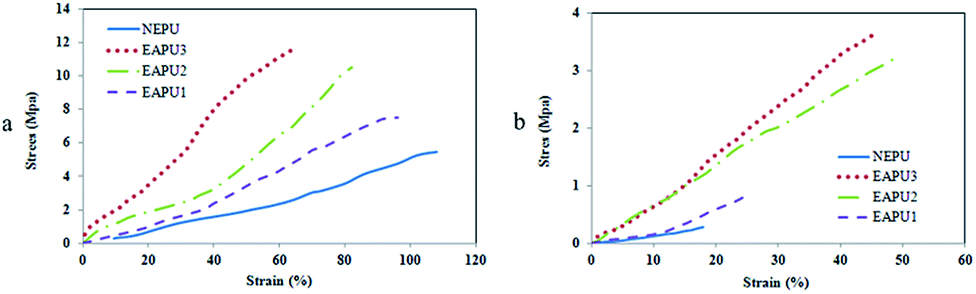 | ||
| Fig. 2 Stress–strain curves for the prepared membranes (a) dry and (b) hydrated states. | ||
As expected, the tensile strength of membranes was reduced under wet condition due to plasticizing effect of the absorbed water molecules. Samples with higher concentration of Si-AT (EAPU2, EAPU3) showed lower drop in tensile strength under wet condition in comparison to NEPU and EAPU1 as a result of the reinforcing effect of AT molecules. Another reason for higher tensile strength for EAPU2 and EAPU3 was lower water absorption of these samples (Table 2) and consequently, lower deterioration of intermolecular interactions due to presence of water molecules between the chain segments. The range of tensile properties and elongation-at-break values in this study implied that most of the prepared membranes were considered suitable materials for wound dressings with appropriate level of mechanical properties.
| Sample | EWA (%) | EWC (%) | Contact angle (°) | WVTR (g/10 cm2 per day) | |
|---|---|---|---|---|---|
| Dry | Wet | ||||
| a According to analysis of variances P-values of <0.05 were considered significant. The difference between quantities with similar superscripts is not significant (p ≥ 0.05) for data of each column. | |||||
| NEPU | 299.3 ± 2.3a | 196.6 ± 4.5a | 59.2 ± 2.3a | 1.15 ± 0.03a | 10.3 ± 0.21a |
| EAPU1 | 277.6 ± 2.5b | 178 ± 2.1b | 61.3 ± 2.1a,b | 1.01 ± 0.02b | 9.8 ± 0.11b |
| EAPU2 | 242.6 ± 7.6c | 141.3 ± 5.8c | 68.4 ± 1.2c | 0.92 ± 0.02c | 9.1 ± 0.22c |
| EAPU3 | 217 ± 1.0d | 116.9 ± 0.5d | 73 ± 1.5d | 0.84 ± 0.01d | 8.4 ± 0.18d |
Hydrophilicity and water fluid handling capacity of wound dressing membranes
The fluid handling capacity is one of the important characteristic of a wound dressing material for maintaining moist environment over wound bed and subsequently preventing scab and scar formation on wounded area.2,40 To achieve this feature, the ideal wound dressing material should maintain balanced hydrophilicity, i.e. they must absorb exudates and toxic components from the wounds bed. Also dressing should regulate evaporative water loss from the wound area at an optimal rate. These features of dressings prevent excessive dehydration and buildup of exudates. In fact, the moist environment facilitates the rapid movement of epithelial cell across wounded skin and also decreases surface inflammation.2,46To evaluate the ability of the prepared membranes for absorbing and retaining wound exudates, their EWA and EWC were measured (Table 2). It was found that NEPU membrane have EWA and EWC around 300% and 200%, respectively. High water absorption ability of this membrane is attributed to the hydrogen bonding type interaction of PEG segment with water molecules. Electroactive membranes with embedded AT moieties showed lower EWA and EWC compared to the NEPU. Sample EAPU3 with highest concentration of Si-AT showed the lowest EWA of EAPU. This behaviour was related to the hydrophobic nature of aromatic AT molecules. Comparison of recorded data with some of important commercial film dressings such as Tegaderm (3 M), Bioclusive (Johnson & Johnson) and Op Site (Smith & Nephew) with EWA of 21, 24 and 35, respectively,46 showed superior performance of the membranes prepared in this study.
The same trend was observed for the surface hydrophilicity of membranes as determined by measuring the contact angle of water droplets on the surface of samples. As shown on Table 2, by increasing the hydrophobic Si-AT content, the contact angle increased from 59° to 73°. Previous studies revealed that the fibroblast cells (as one of important cells that have critical role in wound healing process) show maximum adhesion to the surface of materials with contact angle in the range of 60° and 80°.47 Therefore, the prepared membranes have suitable surface hydrophilicity for effective contribution in cellular activity.
To prevent excessive dehydration as well as build up of exudates, the ideal wound dressing should have adequate WVTR. Commonly used dressings have fixed WVTR regardless of exudate level. Not only different wounds have different exudate levels, but they may vary with time for a specific type of wound. It is expected that modern dressings should have the ability to respond to a wounds exudate level and raise or lower their WVTR to maintain a constant moist environment over wound bed. To estimate the ability of dressing material for proper handling of wound exudates, a test methodology was developed, which involved direct and indirect fluid contact with the dressings.48 The data of WVTR for the prepared membranes are summarized in Table 2.
In dry state, when membranes are not in contact with fluid but just exposed to vapour the NEPU membrane showed higher WVTR than those samples containing Si-AT. This phenomenon can be attributed to either hydrophobic nature of AT segments, or barrier effect of π–π stacked AT moieties against water molecules. The recorded values are much higher than some of commercially available membrane dressings made from dense polyurethane films such as Bioclusive (Johnson–Johnson, 0.394 g/10 cm2 per day), Tegaderm (3 M, 0.492 g/10 cm2 per day) and Op Site (Smith & Nephew, 0.792 g/10 cm2 per day).46 In hydrated state, when membranes are in contact with fluid, much higher values may be recorded for the prepared membranes. The higher WVTR of membranes at wet state can be attributed to the association of chains with water molecules creating structures that facilitate water transportation across the dressing membrane. In fact, by structural expansion or contraction due to changing moisture level the prepared membranes could be able to control their WVTR. This intelligent ability of the prepared materials for self-adjusting of their WVTR in response to exudate level of wound enables them to be applied for management of exudates over a wide range of wounds and different stages during healing process.
To find better perspective regarding practical applicability of the prepared dressing membranes for different wound types and comparison with commercially available dressings another parameter, fluid handling capacity, was calculated (Fig. 3).49,50 This parameter constituted both AC and WVTR and provided better view for exudates managing power of dressings.
 | ||
| Fig. 3 FHC of the membranes at (a) dry and (b) wet states; (c) exudates produced by different wound types (data were collected from ref. 50). (According to analysis of variances P-values of <0.05 were considered significant. The difference between quantities with similar superscripts is not significant (p ≥ 0.05)). | ||
In dry state the calculated FHC values for the prepared membranes were in the range of 1.51 to 1.93 g/10 cm2/24 h. The recorded data was comparable with some of related commercially available thin absorbent wound dressing such as BeneHold (1.97 g/10 cm2/24),51 and hydrocolloid dressings such as Askina Biofilm Transparent (0.51 g/10 cm2/24), Granuflex (1.78 g/10 cm2/24), Algoplaque (1.54 g/10 cm2/24) and Varihesive E (0.51 g/10 cm2/24).52 In hydrated state the FHC values of dressings were increased up to 11.1g/10 cm2/24 h. For comparison, data for some related commercial dressings were as follows: ActivHeal dressing (FHC = 4.6 g/10 cm2/24 h), Allevyn dressing (FHC = 16 g/10 cm2/24 h).50
The intelligent behaviour of the prepared dressing suggests the possible application of these materials for various types of wounds with different levels of exudates (Fig. 3c). These materials can prevent dryness of tissue over lightly exuding wound, and also prevents wound maceration due to accumulation of exudates over heavily exuding wounds.
Preparation of EAPU/nanosilver composites
Utilizing wound dressing membranes with promising antibacterial activity boosts their performance towards protection of wounded area from bacteria which may consequently provide faster healing. There are numerous reports regarding ability of polyaniline and its oligomers for reducing silver ion to its metallic silver form.19,20,53 In the present study, this ability was used for the in situ production of Ag NPs within the prepared membranes. This task was simply performed by immersion of precast membranes in a silver nitrate solution. The redox reaction of AT moieties with Ag+ ions produced Ag NPs. The occurrence of this reaction was studied for EAPU2 sample (representative example). The silver loaded EAPU2 sample was placed in distilled water for 72 h in order to release the produced Ag NPs from the swollen membrane into the water. Then, the aqueous solution containing Ag NPs was subjected to UV-vis spectroscopy measurement. The UV-vis spectrum (Fig. S5†) showed a broad surface plasmon resonance band at about λ = 420 nm. The position and shape of the plasmon absorption depends on the particle size and shape, and the dielectric constant of the surrounding medium. The recorded peak for released Ag NPs was typical of the surface plasmon absorption band of spherical or roughly spherical Ag NPs.54The size of Ag NPs was also measured, since it has determining effect on antibacterial activity.55 For this purpose, the extracted solution containing Ag NPs was deposited on a freshly cleaved Mica film and the AFM analysis was performed on the film after slow evaporation of water. The visualized Ag NPs (Fig. 4) had an average size at about 45 nm. Furthermore, the AFM micrograph exposed the homogeneous size distribution of these nanoparticles.
 | ||
| Fig. 4 AFM micrograph of an in situ generated Ag NPs into EAPU2 (EAPU2-Ag sample). | ||
The weight percent and distribution of Ag NPs within the membranes were also estimated via EDX analysis (Fig. S6†). The analysis was carried out on the cross section of each membrane. A good distribution of Ag NPs (red dots) with higher population near the surface of membranes was detected. According to EDX data, the amount of in situ generated Ag NPs was in the range of 0.06–0.46 w%. This value showed dependency to the concentration of chemically embedded AT moieties. The overall concentration of Ag NPs was increased by higher redox active AT segment concentration in the formulation of membranes. Inspection of Fig. S6† showed very good distribution of the siloxane network (green dots) throughout the membrane, therefore, an isotropic tensile property was expected for these materials.
Electrochemical and electrical properties of membranes
The electrochemical behaviour of the EAPU2 as a typical example of the prepared membranes was examined by cyclic voltammetry (Fig. S7†). EAPU2 showed two oxidation peaks at 0.40 and 0.72 V. The former was assigned to the transition from leucoemeraldine to emeraldine oxidation state, and the latter was corresponded to the transition from emeraldine to pernigraniline oxidation state of AT segments. Based on this study and the result of UV-visible spectrum of Si-AT, the electroactivity of this membrane was confirmed. Standard four probe technique was utilized to measure electrical conductivity of the membranes. These measurements were conducted on CSA doped samples. The results are collected in Table 3. The NEPU sample without AT moieties was an insulator with electrical conductivity of 6.5 × 10−11 S cm−1, but the electrical conductivity was enhanced up to 1.3 × 10−5 S cm−1, for EAPU3 by incorporation of Si-AT segments. This may be attributed to the proper construction of intricate conductive network at high concentration of AT and better π–π stacking of these moieties in the membranes with higher concentration of Si-AT.56 to find better insight regarding actual conductivity of samples in condition near to wounded area, the conductivity measurements were extended for wet (hydrated) samples. The value of electrical conductivity was enhanced considerably when wet (water swollen) electroactive membranes were subjected to four probe measurement. The effect of water on the electrical properties of doped thin polyaniline films was studied by Lubentsov et al.57 They found that the conductivity was increased when water vapor adsorption took place and they also mentioned that doped polyaniline was found to adsorb greater amount of water as compared with undoped samples. Angelopoulos et al.58 have explained that solvated water molecules reduce electrostatic interactions leading to greater charge delocalization and thus increasing conductivity. Therefore, the observed increase in the conductivity of the prepared membranes in hydrated state is in accordance to literature data. It is worth to mention that variation of temperature can also influence on conductivity of polyaniline and other conductive polymers due to possible rearrangement or degradation of their chains.59 To expel this effect, the constant temperature of 30 °C was considered in all conductivity measurements. After in situ production of Ag NPs the value of electrical conductivity was increased. Similarly, this parameter was further increased for the samples containing higher concentration of Si-AT. Again, this phenomenon was attributed to the higher concentration of Ag NPs due to the presence of higher redox active AT moieties. Since microcurrent intensity in the human body is quite low, therefore, this level of conductivity is adequate for transfer of in vivo bioelectrical signals and stimulates cell proliferation and differentiation.60| Sample | Conductivity in dry state (S cm−1) | Conductivity in wet state (S cm−1) |
|---|---|---|
| NEPU | 6.5 × 10−11 | — |
| Doped EAPU1 | 2.6 × 10−7 | 2.7 × 10−3 |
| Doped EAPU2 | 4.5 × 10−6 | 8.5 × 10−3 |
| Doped EAPU3 | 1.3 × 10−5 | 1.2 × 10−2 |
| NEPU-Ag | 6.7 × 10−11 | — |
| EAPU1-Ag | 3.4 × 10−7 | 3.4 × 10−3 |
| EAPU2-Ag | 7.8 × 10−6 | 1.1 × 10−2 |
| EAPU3-Ag | 3.5 × 10−5 | 4.5 × 10−2 |
In vitro cytocompatibility assessment and quantification of cell proliferation
To estimate possible cytotoxicity of membranes, visual inspection of directly contacted L929 cells morphology was performed. The related optical image of L929 fibroblasts cells located at the interface of samples (NEPU, undoped and doped EAPU2, doped EAPU3 and doped EAPU2/Ag) are shown in Fig. 5. No pronounced cell debris and changes in morphology, such as cell lysis and loss of spindle shape occurred which were sufficient evidence the non-cytotoxic behaviour of the prepared membranes. Fortunately, the non-toxic behaviour of sample was also extended for Ag NPs containing membrane (Fig. 5f). Therefore, the concentration of inherently toxic Ag NPs (about 0.3 wt%) produced in the bulk and surface of membranes was appropriate to maintain suitable cytocompatibility against fibroblast cells. | ||
| Fig. 5 Optical microscopy of L-929 cells in direct contact with the membranes (a) negative control (b) NEPU (c) EAPU2 (d) doped EAPU2 9 (e) doped EAPU3 and 9 (f) EAPU2-Ag. | ||
To further confirm this phenomenon, the MTT assay was performed on leachates extracted from membranes after 3 days. Results are collected in Fig. 6. Cells remained viable in contact with extracted leachates from membranes without Ag NPs. Due to the absence of labile linkages in the structure of the prepared membranes; there was no chance for the chemical bond cleavage and consequently, release of possibly toxic materials during the time scale of using such materials for wound dressing application. As well, the purification procedures applied for treatment of final networks (solvent extraction) was successful for expelling any possible residues may remain during preparation of dressing membranes.
 | ||
| Fig. 6 MTT assay for L929 fibroblast cells in contact with leachates extracted from membranes. (According to analysis of variances P-values of <0.05 were considered significant. The difference between quantities with similar superscripts is not significant (p ≥ 0.05)). | ||
After incorporation of Ag NPs in the EAPU membranes, a very low reduction in L929 cells viability was detected that was associated to the toxic nature of the silver nanoparticles released from the membranes. Optimization of Ag NPs concentration has prime importance in the present work since high concentration of these particles can directly improve the antimicrobial activity, but at the same time it can reduce the viability of cells. Fortunately, the reduction in cells viability for EAPU1-Ag and EAPU2-Ag samples was not very high, but still these samples showed perfect antimicrobial property (discussed in next section). In contrast, the viability of cells was significantly reduced for EAPU3-Ag in comparison to two other silver containing samples. In this sample high concentration of AT moieties led to higher in situ reduction of silver ions to Ag NPs. Although such higher Ag NPs may be advantageous for their antimicrobial activity but living cells cannot also tolerate such high concentration. Therefore, this sample was not suitable for the intended application as wound dressing membrane. Fortunately, the versatility of our methodology enable us to tune the desired level of AT and consequently the Ag NPs concentration.
Affinity of cells for adhesion to the biomaterial surface is an excellent indication for evaluation of cytocompatibility of biomaterials. Due to dark colour of the prepared electroactive membranes, it was impossible to follow adhered cells by direct observation of samples surface by optical microscopy. For quantification of cells populated on the prepared membranes, the membranes were cultured with fibroblast cells (type of cell that engage in synthesis of extracellular matrix and collagen and plays a critical role in wound healing process). After certain time intervals (1, 3 and 5 days) the samples were removed from culture media, washed and placed into the new culture media with no cells. Then, the numbers of adhered cells were quantified via MTT assy. Results are collected in Fig. 7.
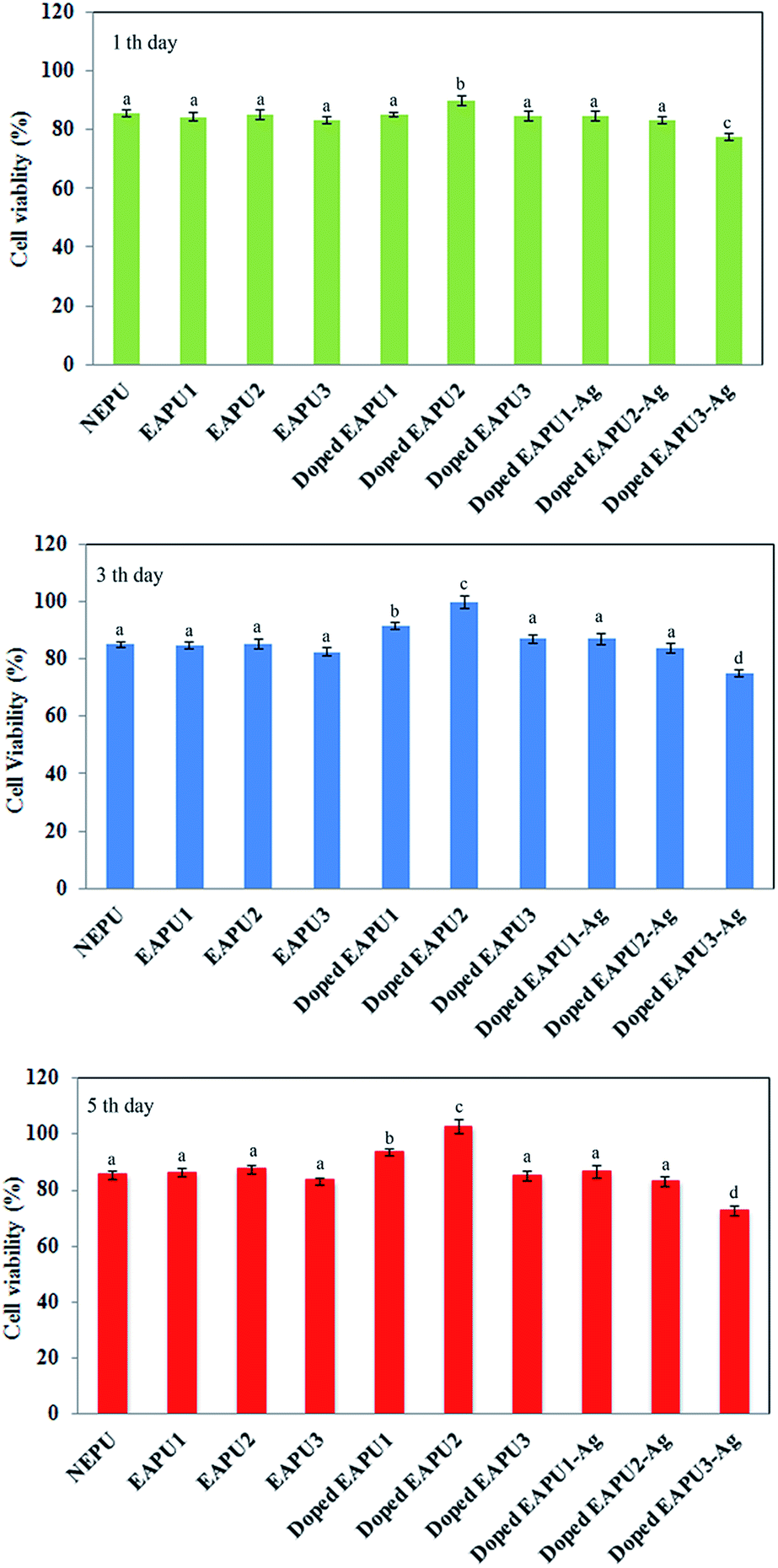 | ||
| Fig. 7 Proliferation of L929 fibroblast cells cultured on the surface of the films for 1, 3 and 5 days. (According to analysis of variances P-values of <0.05 were considered significant. The difference between quantities with similar superscripts is not significant (p ≥ 0.05)). | ||
As it was depicted in Fig. 7, after 1 day culture time, there was no significant difference between non-electroactive and electroactive membranes prior to doping with CSA. However, there was a significant increase (p < 0.05) in the numbers of adhered cells onto the doped EAPU2. With increasing culture time to 3 and 5 days a distinct increase (p < 0.05) was detected for number of adhered and proliferated cells to doped electroactive samples (EAPU1 and EAPU2) in comparison to either non-electroactive, electroactive samples in undoped form and doped EAPU3 with highest concentration of AT moieties. It seems that after attachment of cells to the doped elctroconductive membranes with proper concentration of AT moieties, they showed greater tendency for entering into a rapid proliferative growth phase, establish critical cell–cell interactions and grow to confluence on the membranes. In fact, after doping with CSA, AT moieties of EAPU membranes were able to display their electroactivity and induced the chemical and energy exchange between the cells and its surroundings, which benefit the cell growth. It is well known that charge can influence on the adsorption of proteins and consequently affect cell viability and cell proliferation.61 Meanwhile, toxicity may also develop by introduction of charge on the material. As it was shown in Fig. 7, the developed charge on AT moieties of membranes after doping with CSA exhibited a strong influence on cell growth. The proper concentration of AT moieties in EAPU1 and EAPU2 led to excellent proliferative cell growth as good as tissue culture plate (TCP). The high concentration of AT in doped EAPU3 resulted in reduction in cell viability; however, the cell viability on this sample is still comparable with NEPU and EAPU1-3 in undoped state. As expected, introduction of Ag NPs in surface and bulk of membranes led to slight reduction of cells viability. The EAPU1-Ag and EAPU2-Ag with proper concentration of Ag NPs still showed acceptable cell viability and could support cell growth and proliferation.
For finding better insight regarding the state of adhered cells, the morphology of stained fibroblast cells cultured for 3 days on different membranes was investigated by fluorescence microscopy. The results are collected in Fig. S8.† Fortunately, most of the fibroblast cells were grown properly. They were flat and elongated with spindle-shaped morphology on the surface of the prepared membranes which confirmed their excellent ability to support cell growth and proliferation.
Based on the above discussion regarding mode of interaction of dressing membranes with fibroblast cells, there is promising sign for positive influence of optimum dressing membranes on accelerated healing of the wounded tissue.
The aforementioned in vitro cytocompatibility studies showed desired properties of these materials. These results confirmed the proper selection of starting materials, and indicated the suitability of reaction conditions selected for the preparation of final networks through the sol–gel process. However, judgments regarding biocompatibility of the prepared dressings were made based on data obtained through in vitro study of fibroblast cells behaviour. Evaluating the interaction of other cells involve in healing of dermal wounds such as Keratinocytes and extending these studies on animal models can help to find deeper insight regarding practical usage of such dressing materials.
Antimicrobial activity of dressing membranes
Having wound dressing membranes with antimicrobial activity was another goal of the present study. As it was mentioned, the polyaniline and its derivatives have promising biocidal activity. Reaction of acidic dopants on the polymer chains with bacteria and electrostatic adherence between the polyaniline macromolecules with bacteria are two suggested mechanistic pathways for bactericidal activity of polyaniline.20 Antimicrobial activity of aniline tetramer embedded membranes was evaluated by quantitative “colony forming count” method. The results are collected in Fig. 8 and Table S2.† Investigation of these results revealed that by introduction of AT moieties into the polyurethane/siloxane network biocidal property was induced in these membranes, since all strains under study were killed with moderate to good efficiency in the range of 80–95 %. The antimicrobial activity was increased by increasing the concentration of AT moieties. To improve the antimicrobial activity and reaching to complete killing ability, the impregnation of antimicrobial additive into the membrane was considered in the next step. Silver is a powerful broad spectrum antimicrobial agent that has been used to control infection of the eyes, burns, acute and chronic wounds. Close inspection of antibacterial dressing materials available in the market shows popularity of using silver based dressing to help prevent wound infections. For example, silver containing wound dressings were found to kill gram-negative bacteria such as P. aeruginosa cultures entirely and gram-positive bacteria S. aureus cultures with slightly lower efficiency.62 It has been shown that there is a differential tolerance for silver between mammalian and microbial cells.10 This differential susceptibility provides the excellent opportunity to control the level of silver in wound environment. Therefore, in designing of versatile silver-containing dressing materials the possibility of controlling the amount of impregnated silver particles has prime importance. As described above, the concentration of generated Ag NPs in the prepared dressing could be easily controlled by the content of Si-AT. This asset allowed proper tuning of two opposing characteristic of Ag NPs (bactericidal activity and cytocompatibility) generated in the bulk and surface of dressing membranes. | ||
| Fig. 8 Antibacterial activity of the prepared membranes against (a) S. aureus, (b) C. albicans, (c) P. aeruginosa and (d) E. coli. | ||
At least three mechanistic pathways were reported for the bactericidal effect of Ag NPs on different bacteria. Ag NPs can join to the surface of the cell membrane and disturb some basic functions, such as permeability and respiration. For this mode of action binding of the Ag NPs onto the bacteria directly depends on their available surface area. Since the smaller particles have a larger available surface area for interaction with bacteria they will exhibit a stronger bactericidal effect. Penetration of Ag NPs through cell wall and damaging bacteria, possibly by interacting with sulphur and phosphorus-containing compounds found in abundance throughout the cell membrane is another mechanistic pathway for bactericidal effect of Ag NPs. The smaller Ag NPs penetrate easer through cell wall. Penetrated particles can also inactivate enzymes, generating hydrogen peroxide and causing bacterial cell death.63 Silver ions released from Ag NPs make an additional contribution to the bactericidal effect since DNA loses its replication ability and denaturation of cellular proteins occur upon Ag+ treatment.64
The simple method developed in the present study enabled us to produce spherical or near spherical Ag NPs in an average size of about 45 nm, which is a suitable dimension for having promising antibacterial properties.65 Results of bactericidal activity of Ag NPs containing dressings are collected in Table S2.† According to the recorded data even for EAPU1-Ag sample with the lowest concentration of AT moieties and consequently the lowest amount of generated Ag NPs, a significant bactericidal activity was detected. It is well documented that Ag NPs have lower bactericidal activity on gram positive bacteria.66 This phenomenon can be explained by the presence of thick peptidoglycan and teichoic acids in the gram-positive bacteria cell wall. With increasing the Ag NPs concentration in EAPU2-Ag and EAPU3-Ag almost complete bactericidal activity was detected for all of strains under study.
From antimicrobial point of view, the doped electroconductive membranes (EAPU1-3) have moderate to good antimicrobial activity. However, from cells viability and consequently cells proliferation point of view which is a good indication for possible participation in accelerating of healing process, these membranes have superior property. Antimicrobial activity could be improved to the almost complete killing ability of microorganisms, simply, via immersion of membranes into the silver salt solution and generation of silver nanoparticles on the surface and bulk of the membranes. However, this was associated with reduction in cells viability and proliferation. These broad properties for prepared membranes enabled us to choose the proper dressings based on wound state.
Antioxidant activity of membranes
Inflammation, granulation and remodeling are three stages of wound healing process. In fact, this process well fits to the definition of the “free radical-induced pathologies.” In the inflammatory stage, free radicals (oxyradicals, such as superoxide, hydroxyl ions, and nitric oxide) are produced in high amounts at the site of wound as a defense mechanism against invading bacteria. However, at high concentrations, free radicals can induce severe tissue damage and even lead to neoplastic transformation decreasing the tissue healing by damages in cellular membranes, DNA, proteins and lipids.67 Fibroblasts may also be killed and skin lipids will be made less flexible by excess amount of free radicals.68 Simultaneously, significant reduction in the antioxidant activity of cells in the wounded region occurs by inactivation of enzymic antioxidants and significant depletion of non-enzymic antioxidants level.68 Overall, the occurrences of these events lead to inhibition of mechanisms responsible for the subsequent stages of the healing process (granulation and remodeling), wounds persist in the inflammatory phase and often remain non-healing for longer time. The role of antioxidants appears to be significant in the successful treatment and management of wounds, since these compounds can normalize the wound healing process.There are various reports concerning preparation of wound dressings impregnated with antioxidants, since the effectiveness of the antioxidants is much higher when they are included into a dressing material.69 Previous studies proved that the aniline and its polymeric analogous have radical scavenging effect.8 Therefore, it was anticipated that the embedded AT moieties of the prepared dressing material as an oligomeric analogous of aniline should also have antioxidant property. To evaluate this property of dressing membranes, the common assessment method based on electron transfer reaction to DPPH radicals was utilized. The behavior of AT containing membranes was compared with NEPU sample (Fig. S9†). No reaction occurred between NEPU sample with DPPH solution, since no change was detected in the intensity of peak at 516 nm associated to DPPH radicals. However, a progressive decrease in intensity of this peak was detected, when CSA doped EAPU samples with increasing amount of AT moieties were tested. This phenomenon could be attributed to the electron transfer or hydrogen atom donation from AT segments to DPPH free radicals. Elimination of these radicals led to reduction of the peak intensity at 516 nm. This decrease in peak intensity was more predominant for samples with higher concentration of AT moieties. The efficiency for DPPH scavenging activity of samples is collected in Table 4. The antioxidant activity of EAPU2-Ag was also measured under the same condition. Fortunately, no considerable reduction in antioxidant activity of this sample was recorded in comparison to the corresponding parent membrane (EAPU2). The small decrease in DPPH scavenging effect of silver-containing membrane may be related to partial conversion of AT segment at emeraldine form to pernigraniline state during redox reactions leading to the production of Ag NPs.
| Sample | DPPH scavenged (%) |
|---|---|
| a According to analysis of variances P-values of <0.05 were considered significant. The difference between quantities with similar superscripts is not significant (p ≥ 0.05) for data of each column. | |
| NEPU | 2.1 ± 0.8a |
| EAPU1 | 36.9 ± 1.5b |
| EAPU2 | 65.4 ± 1.1c |
| EAPU3 | 79.2 ± 2.1d |
| EAPU2-Ag | 56.7 ± 1.9e |
Conclusion
Polyurethane/siloxane wound dressing membranes embedded with electroconductive moieties were prepared through sol–gel reaction of trimethoxysilane functional polyurethane prepolymer and aniline tetramer. Presence of PEG segments in polyurethane backbone with high ability for interaction with water molecules offered excellent water absorption capacity for membranes. Meanwhile, existence of polysiloxane domains connecting polyurethane chains together, offered very good dimensional stability even at highly hydrated state. The recorded FHC data revealed smart response of dressing according to the wound exudates level, as the FHC value was up to 10 g/10 cm2 per day at hydrated state and about 2 g/10 cm2 per day at dry state. In fact, the hydrated membranes showed higher tendency for transport of moisture, in comparison to dry membranes. This feature provided facilities for management of wound exudates during different stage of healing process for both heavily and lightly exuding wounds. Tensile strength in the range of 0.3–3.7 MPa at hydrate state confirmed acceptable physical protecting ability of dressing membranes against external trauma.Fibroblast cells viability and proliferation rate were significantly improved for dressing membranes containing proper concentration of AT moieties. Meanwhile, these samples showed moderate to good biocidal activity for gram positive, gram negative and fungal strains. Sample EAPU2 showed the best biological properties, i.e. highest cell compatibility and strongest antimicrobial activity. Introduction of Ag NPs into the dressing membranes simply through immersion of membranes into the silver salt solution offered perfect antimicrobial activity; however, partial reduction in cytocompatibility was detected. Sample EAPU2-Ag showed the balanced properties among silver containing membranes.
All of the electroconductive membranes (with and without Ag NPs) showed promising radical scavenging property, therefore, it is expected that these membranes can participate in regulating the concentration of reactive radical species present in wound site during inflammatory stage of healing process.
In vivo evaluation of selected dressing membranes is under consideration in our lab.
Notes and references
- J. Boateng and K. Matthews, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- C. K. Field and M. D. Kerstein, Am. J. Surg., 1994, 167, S2–S6 CrossRef.
- I. R. Sweeney, M. Miraftab and G. Collyer, Int. Wound J., 2012, 9, 601–612 CrossRef PubMed.
- J. Choi, J. Jang, W. Jang, J. Kim and I. Bae, Biomaterials, 2012, 33, 8579–8590 CrossRef CAS PubMed.
- S. Shimizu, H. Tanaka, S. Sakaki, T. Yukioka, H. Matsuda and S. Shimazaki, J. Trauma, 2002, 52, 683–687 CrossRef CAS PubMed.
- B. E. Britigan, T. L. Roeder, G. T. Rasmussen, D. M. Shasby, M. L. McCormick and C. D. Cox, J. Clin. Invest., 1992, 90, 2187–2196 CrossRef CAS PubMed.
- K. Arunachalam and T. Parimelazhagan, J. Ethnopharmacol., 2013, 145, 139–145 CrossRef CAS PubMed.
- M. Gizdavic-Nikolaidis, J. Travas-Sejdic, P. A. Kilmartin, G. A. Bowmaker and R. P. Cooney, Curr. Appl. Phys., 2004, 4, 343–346 CrossRef PubMed.
- J. P. Saikia, S. Banerjee, B. K. Konwar and A. Kumar, Colloids Surf., B, 2010, 81, 158–164 CrossRef CAS PubMed.
- B. S. Atiyeh, M. Costagliola, S. N. Hayek and S. A. Dibo, Burns, 2007, 33, 139–148 CrossRef PubMed.
- M. R. Gizdavic-Nikolaidis, J. R. Bennett, S. Swift, A. J. Easteal and M. Ambrose, Acta Biomater., 2011, 7, 4204–4209 CrossRef CAS PubMed.
- M. R. Gizdavic-Nikolaidis, J. Bennett, Z. Zujovic, S. Swift and G. a. Bowmaker, Synth. Met., 2012, 162, 1114–1119 CrossRef CAS PubMed.
- S. A. Kramer, J. Vasc. Nurs., 1999, 17, 17–23 CrossRef CAS.
- A. Yari, H. Yeganeh, H. Bakhshi and R. Gharibi, J. Biomed. Mater. Res., Part A, 2014, 102, 84–96 CrossRef PubMed.
- A. Yari, H. Yeganeh and H. Bakhshi, J. Mater. Sci.: Mater. Med., 2012, 23, 2187–2202 CrossRef CAS PubMed.
- P. T. S. Kumar, V.-K. Lakshmanan, T. V. Anilkumar, C. Ramya, P. Reshmi, A. G. Unnikrishnan, S. V. Nair and R. Jayakumar, ACS Appl. Mater. Interfaces, 2012, 4, 2618–2629 Search PubMed.
- N. Shanmugasundaram, T. S. Uma, T. S. Ramyaa Lakshmi and M. Babu, J. Biomed. Mater. Res., Part A, 2009, 89, 472–482 CrossRef CAS PubMed.
- B. Bhowmick, D. Mondal, D. Maity, M. M. Rahaman Mollick, M. Kanti Bain, N. Kumar Bera, D. Rana, S. Chattopadhyay and D. Chattopadhyay, J. Appl. Polym. Sci., 2013, 129, 3551–3557 CrossRef CAS.
- D. Chao, L. Cui, J. Zhang, X. Liu, Y. Li, W. Zhang and C. Wang, Synth. Met., 2009, 159, 537–540 CrossRef CAS PubMed.
- Z. Kucekova, V. Kasparkova, P. Humpolicek, P. Sevcikova and J. Stejskal, Chem. Pap., 2013, 67, 1103–1108 CrossRef CAS.
- L. C. Kloth, Int. J. Low. Extrem. Wounds, 2005, 4, 23–44 CrossRef PubMed.
- G. Shi, Z. Zhang and M. Rouabhia, Biomaterials, 2008, 29, 3792–3798 CrossRef CAS PubMed.
- J. Hu, L. Huang, X. Zhuang, P. Zhang, L. Lang, X. Chen, Y. Wei and X. Jing, Biomacromolecules, 2008, 9, 2637–2644 CrossRef CAS PubMed.
- B. Guo, A. Finne-Wistrand and A.-C. Albertsson, Biomacromolecules, 2010, 11, 855–863 CrossRef CAS PubMed.
- B. Guo, A. Finne-Wistrand and A.-C. Albertsson, Macromolecules, 2011, 44, 5227–5236 CrossRef CAS.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Acta Biomater., 2014, 10, 2341–2353 CrossRef CAS PubMed.
- X. Ma, J. Ge, Y. Li, B. Guo and P. X. Ma, RSC Adv., 2014, 4, 13652 RSC.
- L. Huang, X. Zhuang, J. Hu, L. Lang, P. Zhang, Y. Wang, X. Chen, Y. Wei and X. Jing, Biomacromolecules, 2008, 9, 850–858 CrossRef CAS PubMed.
- Y. Liu, J. Hu, X. Zhuang, P. Zhang, Y. Wei, X. Wang and X. Chen, Macromol. Biosci., 2012, 12, 241–250 CrossRef CAS PubMed.
- H. Zhao, B. Zhu, J. Sekine, S.-C. Luo and H. Yu, ACS Appl. Mater. Interfaces, 2012, 4, 680–686 CAS.
- Y. Liu, H. Cui, X. Zhuang, P. Zhang, Y. Cui, X. Wang, Y. Wei and X. Chen, Macromol. Biosci., 2013, 13, 356–365 CrossRef CAS PubMed.
- H. Cui, Y. Liu, M. Deng, X. Pang, P. Zhang, X. Wang, X. Chen and Y. Wei, Biomacromolecules, 2012, 13, 2881–2889 CrossRef CAS PubMed.
- Y. Liu, H. Cui, X. Zhuang, Y. Wei and X. Chen, Acta Biomater., 2014, 1–7 Search PubMed.
- D. D. Ateh, A. Waterworth, D. Walker, B. H. Brown, H. Navsaria and P. Vadgama, J. Biomed. Mater. Res., Part A, 2007, 83, 391–400 CrossRef CAS PubMed.
- D. D. Ateh, P. Vadgama and H. A. Navsaria, Tissue Eng., 2006, 12, 645–655 CrossRef CAS PubMed.
- S. I. Jeong, I. D. Jun, M. J. Choi, Y. C. Nho, Y. M. Lee and H. Shin, Macromol. Biosci., 2008, 8, 627–637 CrossRef CAS PubMed.
- B. Guo, A. Finne-Wistrand and A.-C. Albertsson, Biomacromolecules, 2011, 12, 2601–2609 CrossRef CAS PubMed.
- A. Serpen, E. Capuano, V. Fogliano and V. Gökmen, J. Agric. Food Chem., 2007, 55, 7676–7681 CrossRef CAS PubMed.
- H. Xu, J. Chang, Y. Chen, H. Fan and B. Shi, J. Mater. Sci., 2013, 48, 6625–6639 CrossRef CAS.
- H.-J. Yoo and H.-D. Kim, J. Biomed. Mater. Res., Part B, 2008, 85, 326–333 CrossRef PubMed.
- M. Alexandru, M. Cazacu, M. Cristea, A. Nistor, C. Grigoras and B. C. Simionescu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1708–1718 CrossRef CAS.
- H. T. Jeon, M. K. Jang, B. K. Kim and K. H. Kim, Colloids Surf., A, 2007, 302, 559–567 CrossRef CAS PubMed.
- D. K. Chattopadhyay, B. Sreedhar and K. V. S. N. Raju, Ind. Eng. Chem. Res., 2005, 44, 1772–1779 CrossRef CAS.
- H. Do Kim and T. W. Kim, J. Appl. Polym. Sci., 1998, 67, 2153–2162 CrossRef.
- Z. Yang, X. Wang, Y. Yang, Y. Liao, Y. Wei and X. Xie, Langmuir, 2010, 26, 9386–9392 CrossRef CAS PubMed.
- H.-J. Yoo and H.-D. Kim, J. Appl. Polym. Sci., 2008, 107, 331–338 CrossRef CAS.
- Y. Tamada and Y. Ikada, J. Colloid Interface Sci., 1993, 155, 334–339 CrossRef CAS.
- S. Palamand, a. M. Reed and L. J. Weimann, J. Biomater. Appl., 1992, 6, 198–215 CrossRef CAS PubMed.
- L.-O. Lamke, G. E. Nilsson and H. L. Reithner, Burns, 1977, 3, 159–165 CrossRef.
- http://www.dressings.org/TechnicalPublications/PDF/S+N-Allevyn-March-2007.
- http://vancive.averydennison.com/en/home/technologies/absorbent-wound-dressing.html.
- http://www.worldwidewounds.com/1997/july/Thomas-Hydronet/hydronet.html.
- C. M. Correa, R. Faez, M. A. Bizeto and F. F. Camilo, RSC Adv., 2012, 2, 3088 RSC.
- J. J. Mock, M. Barbic, D. R. Smith, D. A. Schultz and S. Schultz, J. Chem. Phys., 2002, 116, 6755 CrossRef CAS PubMed.
- C. Tang, W. Sun and W. Yan, RSC Adv., 2014, 4, 523 RSC.
- B. Guo, A. Finne-Wistrand and A.-C. Albertsson, Chem. Mater., 2011, 23, 1254–1262 CrossRef CAS.
- B. Z. Lubentsov, O. N. Timofeeva and M. L. Khidekel', Synth. Met., 1991, 45, 235–240 CrossRef CAS.
- M. Angelopoulos, A. Ray, A. G. Macdiarmid and A. J. Epstein, Synth. Met., 1987, 21, 21–30 CrossRef CAS.
- E. Zampetti, A. Muzyczuk, A. Macagnano, S. Pantalei, S. Scalese, C. Spinella and a. Bearzotti, J. Nanopart. Res., 2011, 13, 6193–6200 CrossRef CAS.
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS PubMed.
- J. Tan, R. A. Gemeinhart, M. Ma and W. M. Saltzman, Biomaterials, 2005, 26, 3663–3671 CrossRef CAS PubMed.
- S.-Y. Ong, J. Wu, S. M. Moochhala, M.-H. Tan and J. Lu, Biomaterials, 2008, 29, 4323–4332 CrossRef CAS PubMed.
- M. Raffi, F. Hussain and T. Bhatti, J. Mater. Sci. Technol., 2008, 24, 192–196 CAS.
- Q. Feng, J. Wu and G. Chen, J. Biomed. Mater. Res., 2000, 52, 662–668 CrossRef CAS.
- M. Guzman, J. Dille and S. Godet, Nanomedicine, 2012, 8, 37–45 CrossRef CAS PubMed.
- N. Baheiraei, F. Moztarzadeh and M. Hedayati, Ceram. Int., 2012, 38, 2921–2925 CrossRef CAS PubMed.
- B. Halliwell, J. M. Gutteridge and C. E. Cross, J. Lab. Clin. Med., 1992, 119, 598–620 CAS.
- I. Süntar, E. K. Akkol, L. Nahar and S. D. Sarker, Free Radicals Antioxid., 2012, 2, 1–7 CrossRef.
- C. Gong, Q. Wu, Y. Wang, D. Zhang, F. Luo, X. Zhao, Y. Wei and Z. Qian, Biomaterials, 2013, 34, 6377–6387 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11454j |
| This journal is © The Royal Society of Chemistry 2014 |