
Reversible sensing of aqueous mercury using a rhodamine-appended polyterthiophene network on indium tin oxide substrates†
Abstract
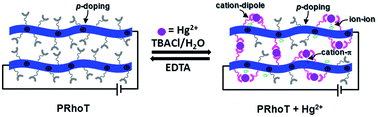
Herein we report the synthesis, and optical and electrochemical properties of a rhodamine-appended polyterthiophene network thin film which demonstrates ion selective potentiometric, chromic and fluorescent responses. The rhodamine-appended terthiophene monomer (RhoT) was electropolymerized and deposited on an ITO electrode. Ion selective potentiometric studies have shown that the potentials of the conducting polymer films decreased upon adding Hg2+ because interactions of Hg2+ with the rhodamine-appended conducting polymer film may increase charge carrier transport properties on a conjugated polymer through rhodamine-bound Hg2+, reduce the doping states by interfering with ions through ion–ion interactions, and perturb the π-extended conjugated polymer through π–π interactions. Moreover, the lower detection limit of the ultrathin film sensor toward Hg2+ (0.10 μM) was less than that obtained from RhoT (1.34 μM), and the response time was less than 30 seconds. Reusability was evaluated by repeating dipping and rinsing cycles in aqueous Hg2+ and EDTA solutions. This approach may provide an easily measurable and inherently sensitive method for Hg2+ ion detection in environmental and biological applications.
 Please wait while we load your content...
Please wait while we load your content...

