
Sonochemical synthesis of highly photoluminescent carbon nanodots†
Kai Wei,
Junjie Li,
Zhishen Ge,
Yezi You and
Hangxun Xu*
CAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei, 230026, Anhui, China. E-mail: hxu@ustc.edu.cn; Fax: +86-551-63607903; Tel: +86-551-63607905
First published on 13th October 2014
Abstract
We present a convenient sonochemical approach for the synthesis of highly photoluminescent carbon nanodots (CDs). CDs were synthesized via pyrolysis of carbon precursors inside implosively collapsing bubbles. We further demonstrate that these CDs can be used for in vitro bioimaging.
Sonochemistry originates from the extreme chemical and physical conditions induced by high intensity ultrasound and has been extensively used in the synthesis of nanostructured materials.1–9 Because acoustic wavelengths are much larger than molecular dimensions, the chemical effects of ultrasound are not derived from direct interactions between ultrasound and chemical species in liquids. Instead, sonochemistry results from the intense compressional heating of gas and vapour inside the collapsing bubbles, and the extraordinary temperatures and pressures thus created (temperatures >5000 K and pressures ∼1000 bar).10
On the other hand, carbon nanodots (CDs) represent an emerging class of photoluminescent nanomaterials with unique optical properties that may have important applications for bioimaging, chemical sensing, and photovoltaic devices.11–19 Since the discovery of CDs, many different synthetic techniques such as arc-discharge, laser-ablation, pyrolysis, solvothermal and microwave heating using various carbon precursors have been developed for the preparation of CDs.20–37 Considering the majority of carbon nanodots (CDs) are prepared using high temperature pyrolysis method to carbonize precursors, we surmise that if appropriate nonvolatile carbon precursors are present in the liquid, the extreme conditions inside collapsing bubbles should be suitable for the pyrolysis of carbon precursors to synthesize CDs.
Here, we demonstrate that a facile sonochemical method can be used to synthesize highly photoluminescent CDs using citric acid as carbon source and ethylenediamine as both a N-doping source and surface passivation agent. In this approach, we employ high intensity ultrasound generated collapsing bubbles saturated with inert gas as microreactors for the pyrolysis of precursor nanodroplets injected (as discussed later) into the interior of bubbles. After solvent evaporation of nanodroplets, citric acid and ethylenediamine can subsequently be carbonized and then form water-soluble CDs. The concept of this sonochemical approach is schematically illustrated in Scheme 1.
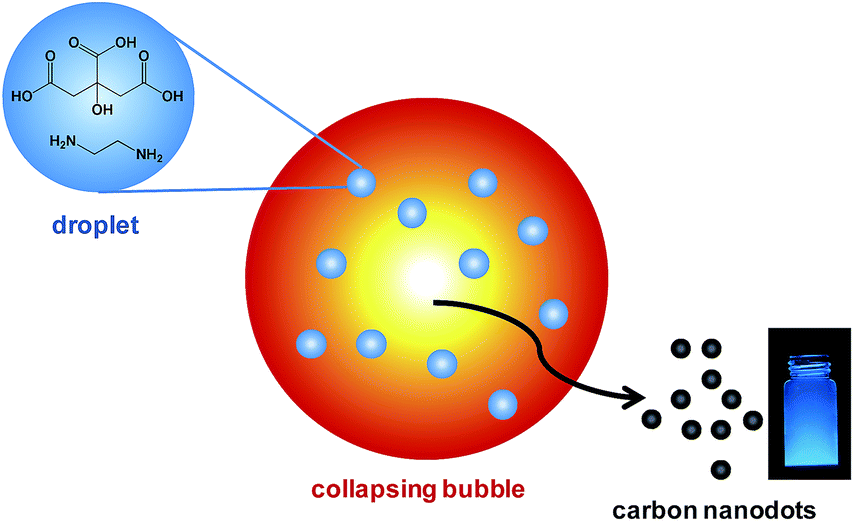 | ||
| Scheme 1 Schematic illustration of using collapsing bubbles generated by high intensity ultrasound as microreactors for the preparation of highly photoluminescent carbon nanodots. The implosively collapsing bubbles can reach extreme conditions sufficient for carbonization of injected carbon precursors. | ||
In a typical experiment, 1 M citric acid was mixed with 1.5 M ethylenediamine (NH2/COOH = 1) and this solution was sparged with Ar for 1 h at 0 °C. Sonication was conducted using an ultrasonic horn (Sonics & Materials, model VCX-500, 1 cm2 Ti horn at 20 kHz and 40 W cm−2) for 8 h under continuous Ar flow. The solution temperature was measured to be 35 °C. The morphology of sonochemically prepared CDs was first characterized by transmission electron microscopy (TEM). As shown in Fig. 1, well-dispersed CDs with diameters of 3–7 nm can be found without apparent aggregation. The sizes of CDs were also measured using atomic force microscopy (AFM), and the average height was around 3 nm (Fig. S1†). Meanwhile, no obvious peaks corresponding to crystalline carbon structures in X-ray diffraction pattern (Fig. S2†) further supported the amorphous nature of CDs.34 This observation is consistent with gas phase sonochemistry usually generates amorphous materials due to the rapid cooling rates (>1010 K s−1).9 13C NMR spectroscopy which can distinguish sp3-hybridized carbon atoms from sp2-hybridized carbon atoms was employed to examine the inherent carbon structures in sonochemically prepared CDs. In the 13C NMR spectrum, signals in the range 30–75 ppm were attributed to aliphatic (sp3) carbons and signals in the range 170–185 ppm were corresponding to carboxyl/amide groups (Fig. S3†).27,38 Elemental analysis revealed the composition of the CDs to be C 37.98 wt%, N 14.46 wt%, H 6.88 wt%, and O 40.68 wt% (calculated).
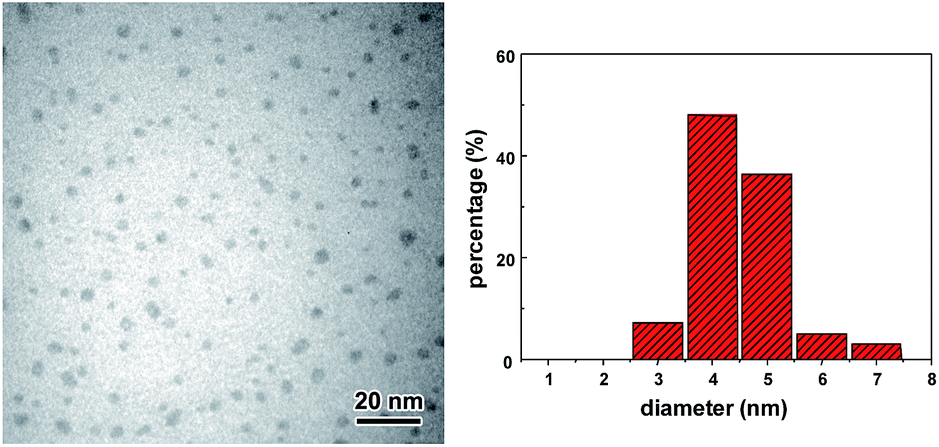 | ||
| Fig. 1 TEM image and corresponding size distribution of the sonochemically synthesized CDs. | ||
The surface structure and component of CDs were identified using Fourier transformed infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS) (Fig. 2). The obtained CDs showed broad absorption band at 3200–3500 cm−1 which can be assigned to stretching vibrations of O–H and N–H. FTIR result also revealed the existence of stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1645 cm−1 and 1730 cm−1 and asymmetric stretching vibration of C–NH–C at 1122 cm−1, which indicated the formation of amide bonds on the surface of CDs.17 Stretching vibration of C–H at 2930 cm−1 and 2870 cm−1 and C–O–C at 1086 cm−1, bending vibrations of N–H at 1557 cm−1 and C–H at 1390 cm−1 were also detected. XPS was further performed to analyze the surface state of the sonochemically prepared CDs and the results showed that they contained primarily carbon, oxygen and nitrogen (Fig. 2b). The high resolution C 1s spectrum (Fig. 2c) showed four peaks at 284.4 eV (C–C), 285.4 eV (C–N), 286.6 eV (C–O) and 287.6 eV (CO).39 The high resolution N 1s spectrum (Fig. 2d) showed two peaks at 399.0 eV (N–H) and 399.8 eV (C–N) which corresponding to amide-N and doping N atoms respectively.34,35 Therefore, primary amine used in this research served dual roles as the N-doping precursors and surface passivation agents. Based on FTIR and XPS results, there are both carboxyl and amino groups on the surface of CDs which make them highly dispersible in aqueous solutions.23,35
O at 1645 cm−1 and 1730 cm−1 and asymmetric stretching vibration of C–NH–C at 1122 cm−1, which indicated the formation of amide bonds on the surface of CDs.17 Stretching vibration of C–H at 2930 cm−1 and 2870 cm−1 and C–O–C at 1086 cm−1, bending vibrations of N–H at 1557 cm−1 and C–H at 1390 cm−1 were also detected. XPS was further performed to analyze the surface state of the sonochemically prepared CDs and the results showed that they contained primarily carbon, oxygen and nitrogen (Fig. 2b). The high resolution C 1s spectrum (Fig. 2c) showed four peaks at 284.4 eV (C–C), 285.4 eV (C–N), 286.6 eV (C–O) and 287.6 eV (CO).39 The high resolution N 1s spectrum (Fig. 2d) showed two peaks at 399.0 eV (N–H) and 399.8 eV (C–N) which corresponding to amide-N and doping N atoms respectively.34,35 Therefore, primary amine used in this research served dual roles as the N-doping precursors and surface passivation agents. Based on FTIR and XPS results, there are both carboxyl and amino groups on the surface of CDs which make them highly dispersible in aqueous solutions.23,35
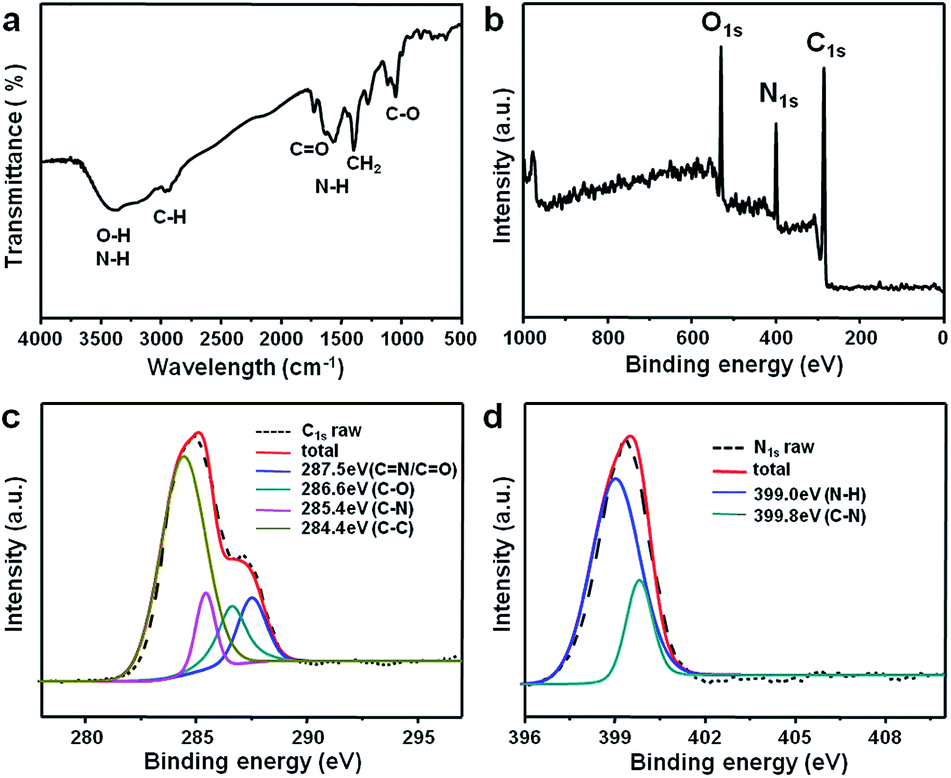 | ||
| Fig. 2 (a) FTIR spectrum, (b) XPS survey scan, (c) C 1s and (d) N 1s spectra of the CDs. | ||
The UV/Vis absorption spectrum showed a peak at 354 nm (Fig. 3a). When these CDs were excited at 360 nm, a maximum emission peak at 450 nm was observed (Fig. 3b). Using quinine sulfate as reference, the quantum yield was measured to reach 44.8%, indicating superior photoluminescence performance to previously reported CDs using similar carbon precursors.34 These CDs exhibited excitation-dependent emission behavior only when the excitation wavelength is larger than 400 nm. The photoluminescent peak shifted from 460 nm to 508 nm when the excitation wavelength changed from 400 nm to 460 nm. In contrast, the emission wavelength remained unchanged at ∼450 nm when the excitation wavelength varied from 320 nm to 400 nm. Although the mechanism for the origin of luminescence from CDs remains controversial, it is generally accepted that different surface states caused by different functional groups are responsible for these photoluminescent properties.40 The emission-independent behavior in the range of 320–400 nm may indicate the uniform surface feature of these sonochemically prepared CDs. The fluorescence lifetime (τ) of CDs was detected by time-correlated single-photo counting (TCSPC). The decay trace was fitted by biexponential function and the average lifetime was calculated about 15.52 ns which contained two components of 3.21 ns (4%) and 16.07 ns (96%) (Fig. S4†).
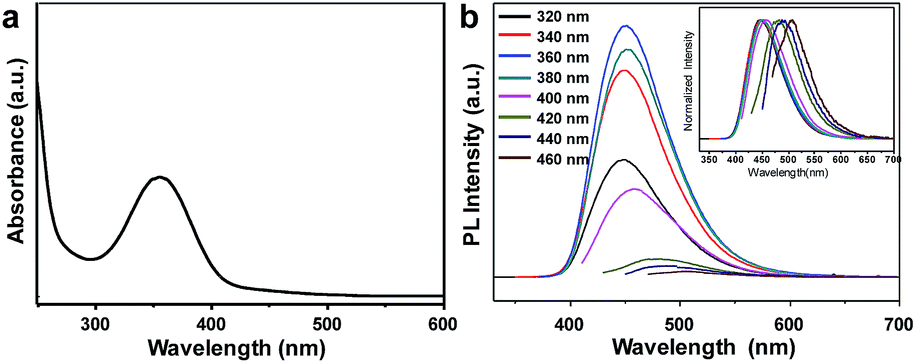 | ||
| Fig. 3 (a) UV/Vis absorption spectrum and (b) photoluminescence emission spectra of the CDs excited at different wavelengths. | ||
The quantum yield of CDs can be tuned from 7.3% to 44.8% by varying the molar ratio of NH2/COOH (Table S1†). The highest quantum yield was obtained when the ratio of NH2/COOH was 1/1, suggesting these two functional groups react with each other to form amide bonds for the photoluminescence enhancement. Sonication time only has a minor influence on the quantum yield of sonochemically prepared CDs (Table S1†). This observation supports that CDs are formed in the gas phase of collapsing bubbles, as solution phase reaction usually leads to the formation of larger nanoparticles when sonication time increases.41 Instead, because CDs are produced by pyrolysis of injected nanodroplets inside collapsing bubbles, lager CDs could be obtained using concentrated precursors. As shown in Fig. S5,† CDs with diameters approximately 6–25 nm can be obtained when the concentration of citric acid was increased to 2 M. They exhibited similar absorption and photoluminescence properties to those CDs synthesized using 1 M citric acid but with lower quantum yield (Table S1†). The quantum yield can reach as high as 77.3% when the concentration of citric acid was adjusted to 0.5 M while the molar ratio of NH2/COOH remained 1/1 (Table S1†). Citric acid was used as sole carbon precursor and the quantum yield of such obtained CDs was only 1.1%. Meanwhile, the absorption peak shifted to 330 nm (Fig. S6†). The presence of primary amine in the precursor solution could dramatically enhance the photoluminescence of CDs by doping CDs with nitrogen and modifying their surfaces.34 Therefore, the photoluminescence of the CDs should be attributed to the radiative recombination of electrons and holes trapped on the CDs surface.29,42–44
As an emerging class of photoluminescent nanomaterial, CDs have attracted increasing attention due to their advantages in stable photoluminescence, tunable emission spectra, low cytotoxicity and excellent biocompatibility.45,46 Because the major component of CDs is the nontoxic element carbon, they are considered to be promising alternatives to semiconductor quantum dots which are usually composed of toxic heavy metals such as cadmium. To assess the potential biological imaging application of sonochemically prepared CDs, HeLa cells were first used to evaluate the cytotoxicity by MTT assay. The viability was above 80% when the concentration of CDs was 100 μg ml−1 which should be sufficient for bioimaging applications. This also indicated that the sonochemically synthesized CDs exhibited low cytotoxicity. Then CDs were cocultured with HeLa cells for in vitro bioimaging using a confocal microscopy. As shown in Fig. 4, the labeled cells showed bright blue luminescence when excited at 405 nm. The CDs were mainly localized in the cell membrane and cytoplasmic area, but barely observable in the nucleus. Although it is likely that they were internalized into the cells by endocytosis, the exact internalization mechanism requires further inverstigations.23,37 The photostability of CDs was also evaluated by comparison to FITC which is a fluorophore commonly used in microscopy (Fig. S7†). CDs and FITC were consistently irradiated by a mercury lamp (4.3 mW cm−2). The photoluminescence intensity of FITC was quenched by 80% in 60 min. In contrast, the photoluminescence intensity of CDs only dropped 20% during the same period due to the surface oxidation reactions caused by UV irradiation. Therefore, these sonochemically synthesize CDs should be suitable for long-term bioimaging.
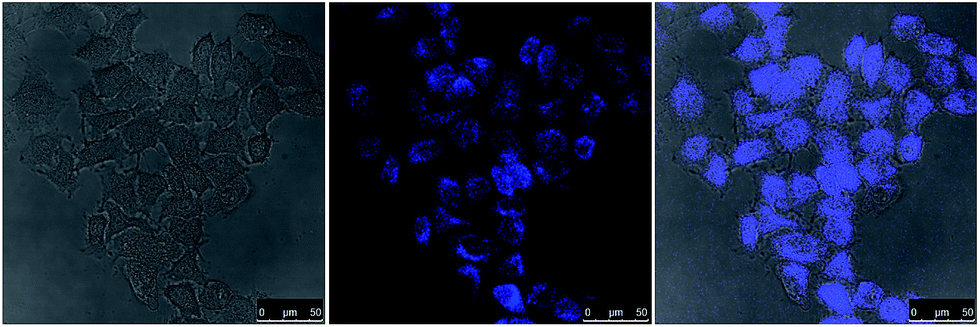 | ||
| Fig. 4 Laser scanning confocal microscopy images of HeLa cells labeled by CDs. The cells were excited by 405 nm laser. The concentration of CDs was 50 μg ml−1. Left: bright-field image; middle: the photoluminescence image; right: superposition of the photoluminescence and bright-field images. | ||
Sonochemistry can be applied to synthesize nanostructured materials from either volatile or nonvolatile precursors and can be categorized as primary sonochemistry and secondary sonochemistry.9 Primary sonochemistry occurs inside collapsing bubbles and usually involves volatile species.47,48 Secondary sonochemistry is a solution-phase chemistry and sonochemical products may arise from chemically active species (e.g., radicals from thermal decomposition of vapour molecules) generated inside bubbles which diffuse into the solution to initiate chemical reactions.49–51 Previously, based on a series of spectroscopic experiments, we have proved that liquid nanodroplets containing nonvolatile species can be injected into the collapsing bubbles by capillary wave action, microjetting, or bubble coalescence because of the significant deformation during bubble collapse in the dense cloud of cavitating bubbles.52,53 Once the nanodroplets enter the hot interior of a collapsing bubble, the solvent evaporates and chemical reactions in the gas phase analogous to the processes in flame pyrolysis occur. Therefore, nonvolatile species can be excited and pyrolyzed inside the collapsing bubbles. Crossing the line between primary and secondary sonochemistry, that is, the application of collapsing bubbles as microreactors to pyrolyze nonvolatile precursors has not been reported yet.
As further evidence of the sonochemical reactions of nonvolatiles, we used polyacrylic acid (PAA, Mw = 1800) to replace citric acid as carbon precursor to synthesize CDs. The CDs show an absorption peak at 348 nm and the photoluminescence spectra exhibit excitation-dependent emission behavior (Fig. S8†). By optimizing the synthetic conditions, the quantum yield can reach 12%. PAA obviously is a nonvolatile carbon source and the pyrolysis of PAA can only occur inside bubbles. Therefore, it further confirms that this sonochemical approach produces CDs via pyrolysis carbon precursors inside the collapsing bubbles.
In conclusion, a facile sonochemical method is developed for the synthesis of highly photoluminescent CDs. The collapsing bubbles generated by high intensity ultrasound act as microreactors and provide extreme, transient conditions suitable for pyrolysis of carbon precursors. By adding primary amines as N-doping sources and surface passivation agents, sonochemically prepared CDs exhibit high quantum yield and excellent photostability.
Acknowledgements
The authors gratefully acknowledge helpful discussions from K. S. Suslick and the funding support from National Natural Science Foundation of China (grant number 51402282 and 21474095), the National Key Basic Research Program of China (2015CB351903) and the Fundamental Research Funds for the Central Universities.Notes and references
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039 CrossRef CAS PubMed.
- D. G. Shchukin, D. Radziuk and H. Mohwald, Annu. Rev. Mater. Res., 2010, 40, 345 CrossRef CAS.
- K. Vinodgopal, B. Neppolian, I. V. Lightcap, F. Grieser, M. Ashokkumar and P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 1987 CrossRef CAS.
- J. Geng, L. P. Jiang and J. J. Zhu, Sci. China: Chem., 2012, 55, 2292 CrossRef CAS.
- S. E. Skrabalak, Phys. Chem. Chem. Phys., 2009, 11, 4930 RSC.
- A. Gedanken, Ultrason. Sonochem., 2004, 11, 47 CrossRef CAS PubMed.
- T. Gao and T. Wang, Chem. Commun., 2004, 2558 RSC.
- P. Hasin and Y. Y. Wu, Chem. Commun., 2012, 48, 1302 RSC.
- H. X. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555 RSC.
- K. S. Suslick and D. J. Flannigan, Annu. Rev. Phys. Chem., 2008, 59, 659 CrossRef CAS PubMed.
- H. T. Li, Z. H. Kang, Y. Liu and S. T. Lee, J. Mater. Chem., 2012, 22, 24230 RSC.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726 CrossRef CAS PubMed.
- S. T. Yang, L. Cao, P. G. Luo, F. S. Lu, X. Wang, H. F. Wang, M. J. Meziani, Y. F. Liu, G. Qi and Y. P. Sun, J. Am. Chem. Soc., 2009, 131, 11308 CrossRef CAS PubMed.
- F. Wang, Y. Chen, C. Liu and D. Ma, Chem. Commun., 2011, 47, 3502 RSC.
- L. Zhou, Y. H. Lin, Z. Z. Huang, J. S. Ren and X. G. Qu, Chem. Commun., 2012, 48, 1147 RSC.
- S. N. Qu, H. Chen, X. M. Zheng, J. S. Cao and X. Y. Liu, Nanoscale, 2013, 5, 5514 RSC.
- W. Kwon, G. Lee, S. Do, T. Joo and S. W. Rhee, Small, 2014, 10, 506 CrossRef CAS PubMed.
- B. Kong, A. W. Zhu, C. Q. Ding, X. M. Zhao, B. Li and Y. Tian, Adv. Mater., 2012, 24, 5844 CrossRef CAS PubMed.
- D. Y. Pan, J. C. Zhang, Z. Li, C. Wu, X. M. Yan and M. H. Wu, Chem. Commun., 2010, 46, 3681 RSC.
- X. Y. Xu, R. Ray, Y. L. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736 CrossRef CAS PubMed.
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. F. Wang, P. G. Luo, H. Yang, M. E. Kose, B. L. Chen, L. M. Veca and S. Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756 CrossRef CAS PubMed.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, V. Georgakilas and E. P. Giannelis, Chem. Mater., 2008, 20, 4539 CrossRef CAS.
- P. C. Hsu and H. T. Chang, Chem. Commun., 2012, 48, 3984 RSC.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473 CrossRef CAS PubMed.
- Y. Yang, J. Cui, M. Zheng, C. Hu, S. Tan, Y. Xiao, Q. Yang and Y. Liu, Chem. Commun., 2012, 48, 380 RSC.
- W. Kwon and S. W. Rhee, Chem. Commun., 2012, 48, 5256 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953 CrossRef CAS PubMed.
- S. Liu, J. Q. Tian, L. Wang, Y. W. Zhang, X. Y. Qin, Y. L. Luo, A. M. Asiri, A. O. Al-Youbi and X. P. Sun, Adv. Mater., 2012, 24, 2037 CrossRef CAS PubMed.
- Y. Q. Dong, H. C. Pang, H. B. Yang, C. X. Guo, J. W. Shao, Y. W. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800 CrossRef CAS PubMed.
- S. W. Yang, J. Sun, X. B. Li, W. Zhou, Z. Y. Wang, P. He, G. Q. Ding, X. M. Xie, Z. H. Kang and M. H. Jiang, J. Mater. Chem. A, 2014, 2, 8660 CAS.
- H. L. Dong, A. Kuzmanoski, D. M. Gobl, R. Popescu, D. Gerthsen and C. Feldmann, Chem. Commun., 2014, 50, 7503 RSC.
- H. T. Li, X. D. He, Y. Liu, H. Huang, S. Y. Lian, S. T. Lee and Z. H. Kang, Carbon, 2011, 49, 605 CrossRef CAS PubMed.
- H. Zhu, X. L. Wang, Y. L. Li, Z. J. Wang, F. Yang and X. R. Yang, Chem. Commun., 2009, 5118 RSC.
- X. Y. Zhai, P. Zhang, C. J. Liu, T. Bai, W. C. Li, L. M. Dai and W. G. Liu, Chem. Commun., 2012, 48, 7955 RSC.
- P. Zhang, W. C. Li, X. Y. Zhai, C. J. Liu, L. M. Dai and W. G. Liu, Chem. Commun., 2012, 48, 10431 RSC.
- C. Z. Zhu, J. F. Zhai and S. J. Dong, Chem. Commun., 2012, 48, 9367 RSC.
- C. Liu, P. Zhang, F. Tian, W. Li, F. Li and W. Liu, J. Mater. Chem., 2011, 21, 13163 RSC.
- S. N. Qu, X. Y. Wang, Q. P. Lu, X. Y. Liu and L. J. Wang, Angew. Chem., Int. Ed., 2012, 51, 12215 CrossRef CAS PubMed.
- W. Kwon, S. Do, J. Lee, S. Hwang, J. K. Kim and S. W. Rhee, Chem. Mater., 2013, 25, 1893 CrossRef CAS.
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng, J. Hao and S. P. Lau, ACS Nano, 2012, 6, 5012 Search PubMed.
- H. X. Xu and K. S. Suslick, ACS Nano, 2010, 4, 3209 CrossRef CAS PubMed.
- L. Bao, Z. Zhang, Z. Tian, L. Zhang, C. Liu, Y. Lin, B. Qi and D. Pang, Adv. Mater., 2011, 23, 5801 CrossRef CAS PubMed.
- H. Zheng, Q. Wang, Y. Long, H. Zhang, X. Huang and R. Zhu, Chem. Commun., 2011, 47, 10650 RSC.
- X. M. Li, S. L. Zhang, S. A. Kulinich and H. B. Zeng, Sci. Rep., 2014, 4, 4976 Search PubMed.
- W. B. Li, Z. Yue, C. Wang, W. Zhang and G. H. Liu, RSC Adv., 2013, 3, 20662 RSC.
- X. F. Hu, L. Cheng, N. Wang, L. Sun, W. Wang and W. G. Liu, RSC Adv., 2014, 4, 18818 RSC.
- S. H. Jeong, J. H. Ko, J. B. Park and W. Park, J. Am. Chem. Soc., 2004, 126, 15982 CrossRef CAS PubMed.
- K. S. Suslick, M. Fang and T. Hyeon, J. Am. Chem. Soc., 1996, 118, 11960 CrossRef CAS.
- R. A. Caruso, M. Ashokkumar and F. Grieser, Langmuir, 2002, 18, 7831 CrossRef CAS.
- S. Anandan, F. Grieser and M. Ashokkumar, J. Phys. Chem. C, 2008, 112, 15102 CAS.
- K. Okitsu, K. Sharyo and R. Nishimura, Langmuir, 2009, 25, 7786 CrossRef CAS PubMed.
- H. X. Xu, N. C. Eddingsass and K. S. Suslick, J. Am. Chem. Soc., 2009, 131, 6060 CrossRef CAS PubMed.
- H. X. Xu, N. G. Glumac and K. S. Suslick, Angew. Chem., Int. Ed., 2010, 49, 1079 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10354h |
| This journal is © The Royal Society of Chemistry 2014 |