
Sustainable joint solventless coproduction of glycerol carbonate and ethylene glycol via thermal transesterification of glycerol†
Abstract
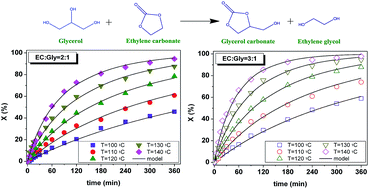
This study focuses on the thermal reaction between glycerol and ethylene carbonate to obtain glycerol carbonate and ethylene glycol under solventless homogeneous operation, the process being a transcarbonation of glycerol or a glycerolysis of ethylene carbonate. As the two reagents constitute an immiscible system at 40 °C evolving into a single phase at 80 °C, the evolution of phases with temperature was studied by focused beam reflectance measurement. As the biphasic system was inert, runs were completed under a monophasic regime from 100 to 140 °C with molar ratios of ethylene carbonate to glycerol of 2 and 3, achieving quantitative conversion of glycerol, as corroborated by a thermodynamic study. Second order potential kinetic models were proposed and fitted to the data. Finally, a comparison with analogous catalytic approaches was made, showing that this process performs better material-wise.
 Please wait while we load your content...
Please wait while we load your content...

