
An efficient D-glucosamine-based copper catalyst for C–X couplings and its application in the synthesis of nilotinib intermediate†
Ming Wena,
Chao Shenb,
Linfang Wanga,
Pengfei Zhang*c and
Jianzhong Jin*ab
aCollege of Petroleum Chemical Industry, Changzhou University, Changzhou 213164, China
bCollege of Biology and Environmental Engineering, Zhejiang Shuren University, Hangzhou 310015, China
cCollege of Material Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou 310036, China. E-mail: chxyzpf@hotmail.com; Fax: +86-571-28862867; Tel: +86-571-28862867
First published on 14th November 2014
Abstract
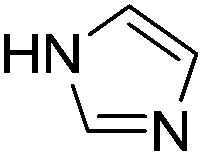
D-Glucosamine has been studied for C–N and C–S bond formations via cross-coupling reactions of nitrogen and sulfur nucleophiles with both aryl iodides and bromides. Imidazoles, benzimidazole, indole, pyrrolidine and diphenyl disulfide undergo reactions with aryl halides in the presence of 10 mol% D-glucosamine, 10 mol% CuI, and 2 equiv. of Cs2CO3 in DMSO–H2O at moderate temperature to give the corresponding products in good to excellent yields. Substrates bearing halides, free amino groups, trifluoromethyl and heterocycles were well tolerated. The high water solubility of the ligand enables easy catalyst removal. In addition, the application of this catalytic system for the synthesis of nilotinib intermediate was also successfully demonstrated using commercially available substrates.
Introduction
The method of building carbon-heteroatom structures has long been a hot topic in the field of organic synthesis because of their wide existence in natural products, which have biological activities, drugs and polymer materials.1–4 During the past few decades, many efforts from different groups have led to the discovery of a wide variety of Pd-catalyzed methods for the formation of carbon-heteroatom bonds.5,6 Although such coupling reactions have high efficiency, the high price of Pd and the higher toxicity of Pd hindered the application of cross-coupling reactions on many occasions. In the context of advocacy of green chemistry in the 21st century, copper catalysts showed attractive advantages, which were cheap and low toxicity.7,8 They increase the applicability of the reaction and avoid the phenomenon of the shift of double bonds, which was caused by the elimination of β-H in catalytic coupling by a Pd catalyst. At present, C–N coupling catalyzed by copper has achieved fruitful results,9–14 but some reactions often suffer from several drawbacks such as the use of stoichiometric copper catalysts, high polarity and toxicity of solvents, harsh reaction conditions, and special ligands, including phenanthrolines,15 amino acids,16 diketones,17 diamines,18 oximes,19 carbohydrates20 and others;21 some of the above conditions not only cause great waste, but also pollute the environment we live in.D-Glucosamine has been selected as a cheap and readily available chiral scaffold for the synthesis of a series of novel ligands and organocatalysts because of its excellent advantages that include green chemistry, environmental protection, economy and efficiency.22,23 The design and fine-tuning of carbohydrate-based ligands are facilitated by the multiple functional groups within this class of compounds.24–27 In preliminary communication,28 we reported the catalysis of chitosan (CS) for the cross-coupling reactions of aryl halides and sodium sulfinates, which provides a simple and extremely efficient new route to unsymmetrical diaryl sulfones. These features led us to further study the scope of D-glucosamine for catalysis of carbon-heteroatom cross-coupling reactions.29
In this paper, we describe an efficient D-glucosamine-based catalytic system for the formations of C–N and C–S bond via cross-coupling reactions of nitrogen and sulfur nucleophiles with both aryl iodides and bromides. Imidazoles, benzimidazole, indole, pyrrolidine and diphenyl disulfide undergo reactions with aryl halides in the presence of 10 mol% CuI and 2 equiv. of Cs2CO3 in DMSO–H2O at moderate temperature using 10 mol% D-glucosamine as a green ligand. In addition, the application of this catalytic system for the synthesis of nilotinib intermediate is also successfully demonstrated using commercially available substrates.
Results and discussion
At first, 4-iodoanisole (1a) and imidazole (2a) were selected as model substrates and a series of natural ligands screened under conditions of 10 mol% CuI, 10 mol% ligand, 2 equiv. of K2CO3 as base and DMSO–H2O (3 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture as solvent at 100 °C for 10 h. D-Glucose L1 was used as ligand first and the desired product 3a was obtained in 75% yield. Chitosan L2 only gave the product in 42% yield due to its poor solubility. Then, other monosaccharide molecules such as D-galactose L3, D-glucosamine L4, L-arabinose L5 and L-galactono-1,4-lactone L6 were investigated (Scheme 1). According to the results in Scheme 1, it was obvious that D-glucosamine was the most efficient ligand examined and it gave the coupling product in 80% yield under air.
:1) mixture as solvent at 100 °C for 10 h. D-Glucose L1 was used as ligand first and the desired product 3a was obtained in 75% yield. Chitosan L2 only gave the product in 42% yield due to its poor solubility. Then, other monosaccharide molecules such as D-galactose L3, D-glucosamine L4, L-arabinose L5 and L-galactono-1,4-lactone L6 were investigated (Scheme 1). According to the results in Scheme 1, it was obvious that D-glucosamine was the most efficient ligand examined and it gave the coupling product in 80% yield under air.
 | ||
| Scheme 1 Ligand comparison in Cu-catalyzed N-arylation of imidazole with 4-iodoanisole (reaction conditions: 4-iodoanisole 1a (1.0 mmol), imidazole 2a (1.2 mmol), CuI (0.1 mmol), ligand (0.1 mmol), K2CO3 (2.0 mmol), DMSO–H2O (3 mL, 1:1), under air, 10 h). | ||
Next, several catalysts, organic solvent/H2O (1:1) mixtures, bases and temperatures were screened for this reaction. With D-glucosamine as ligand, different types of copper sources were screened under other identical conditions. Only 16% yield was obtained without ligand (Table 1, entry 1). Compared with the catalytic effect of CuI, other copper sources, such as CuBr, CuBr2, CuF2 and Cu(OAc)2·H2O, showed slightly lower catalytic activities (Table 1, entries 1–5). No C–N coupling product was obtained when the reaction was carried out with Pd(OAc)2 as catalyst (Table 1, entry 6). The reactions did not lead to the formation of a homocoupled biaryl compound, and the controlled experiments without D-glucosamine showed no reaction (Table 1, entry 7). In addition to DMSO, DMF, THF and CH3CN were further surveyed; nevertheless, these solvents were not good choices here and the yields were comparatively lower (Table 1, entries 8–10). Next, several bases, such as Cs2CO3, KOH, NaOH, Na2CO3 and LiOH, were screened for this coupling reaction. Fortunately, we found that the desired product 3a was obtained in 90% yield with Cs2CO3 as base (Table 1, entry 11). While checking the minimum requirement of catalyst loading for the best performance of the reaction, it has been found that decreasing the catalyst loading will affect the yield of the product. For example, when 5 mol% of L5 was used, only 71% yield was achieved. Finally, the reaction temperature was screened for this reaction. When the reaction was performed at 60 or 80 °C, the reaction was found to be inefficient (Table 1, entries 16 and 17). When the reaction temperature was increased to 120 °C under other identical conditions, the yield was very similar to the former, showing that 100 °C was the optimum reaction temperature (Table 1, entry 18).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Solvent:H2O (1:1) |
Base (2 equiv.) | Temp [°C] | Yieldb (%) |
| a Reaction conditions: 4-iodoanisole 1a (1.0 mmol), imidazole 2a (1.2 mmol), CuI (0.1 mmol), D-glucosamine (0.1 mmol), base (2.0 mmol), solvent: H2O (3.0 mL 1:1), under air, 10 h.b GC-MS yield.c Without ligand.d 5 mol% of D-glucosamine was used. |
|||||
| 1 | CuI | DMSO | K2CO3 | 100 | 80(16)c |
| 2 | CuBr | DMSO | K2CO3 | 100 | 55 |
| 3 | CuBr2 | DMSO | K2CO3 | 100 | 64 |
| 4 | CuF2 | DMSO | K2CO3 | 100 | 54 |
| 5 | Cu(OAc)2·H2O | DMSO | K2CO3 | 100 | 7 |
| 6 | Pd(OAc)2 | DMSO | K2CO3 | 100 | Trace |
| 7 | — | DMSO | K2CO3 | 100 | 0 |
| 8 | CuI | DMF | K2CO3 | 100 | 13 |
| 9 | CuI | THF | K2CO3 | 100 | 14 |
| 10 | CuI | CH3CN | K2CO3 | 100 | Trace |
| 11 | CuI | DMSO | Cs2CO3 | 100 | 90(71)d |
| 12 | CuI | DMSO | KOH | 100 | 67 |
| 13 | CuI | DMSO | NaOH | 100 | 84 |
| 14 | CuI | DMSO | Na2CO3 | 100 | 39 |
| 15 | CuI | DMSO | LiOH | 100 | 55 |
| 16 | CuI | DMSO | Cs2CO3 | 60 | 67 |
| 17 | CuI | DMSO | Cs2CO3 | 80 | 72 |
| 18 | CuI | DMSO | Cs2CO3 | 120 | 88 |

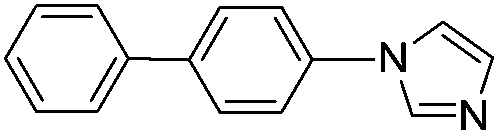
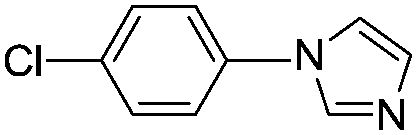
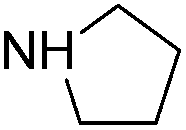
With the optimized reaction conditions in hand (DMSO:H2O (1:1) as solvent, 10 mol% D-glucosamine as ligand, 10 mol% CuI as catalyst, 2 equiv. of Cs2CO3 as base, and completing the reaction at 100 °C under air for 10 h), we began our investigation into the scope of D-glucosamine-catalyzed coupling of aryl halides and nitrogen nucleophiles and the results are summarized in Table 2. The results showed that good yields were obtained for the coupling of aryl iodides with imidazole (Table 2, entries 1–7). In general, aryl iodides bearing electron-withdrawing groups, such as –CF3 and –Cl, were more reactive compared to aryl iodides bearing electron-donating groups, such as –OMe, –Me and –NH2, which afforded the corresponding N-arylated imidazoles in lower yields. For example, the reaction of 1-trifluoromethyl-4-iodobenzene and imidazole proceeded with 87% isolated yield (Table 2, entry 6), whereas the inactive 4-iodoaniline required more than 24 h reaction time to give the corresponding product in 70% isolated yield (Table 2, entry 3). Next, the coupling of imidazole with 4-iodo-pyrazole was examined using the same optimized conditions. 4-Iodo-pyrazole gave the product in 70% isolated yield (Table 2, entry 8). Fortunately, it was found that bromobenzene worked well, affording the corresponding product in satisfactory yield (Table 2, entry 9). However, the N-arylation of imidazole with chlorobenzene was unsuccessful under the same conditions (Table 2, entry 10). We also employed 4-methylimidazole as a nucleophilic reagent, which only gave a moderate yield (Table 2, entry 11). Further experiments with different nitrogen nucleophiles such as indole and pyrrolidine were carried out under these optimized conditions. It was clear that amination proceeded very effectively and afforded the corresponding products 3j and 3k in good yields (Table 2, entries 12 and 13). Considering the development of catalytic methods that selectively produce the N-arylated product from a substrate, which has two nucleophilic nitrogens, (NH and NH2 groups) is still challenging.30 Next, we explored chemoselective C–N coupling using 2-aminobenzimidazole as reaction substrate under our optimal conditions and the corresponding product 3l was obtained in 72% yield (Table 2, entry 14).
|
||||
|---|---|---|---|---|
| Entry | Aryl halides | Nitrogen nucleophile | Product | Yieldb (%) |
| a Reaction conditions: 1 (1.0 mmol), 2 (1.2 mmol), CuI (0.1 mmol), D-glucosamine (0.1 mmol), Cs2CO3 (2.0 mmol), DMSO:H2O (3.0 mL 1:1), under air.b Isolated yield.c 24 h.d No reaction. |
||||
| 1 | 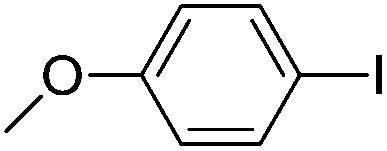 |
 |
 |
84(3a) |
| 2 | 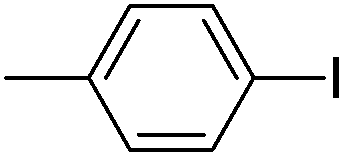 |
 |
 |
81(3b) |
| 3 |  |
 |
 |
70(3c)c |
| 4 |  |
 |
 |
71(3d) |
| 5 |  |
 |
 |
86(3e) |
| 6 |  |
 |
 |
87(3f) |
| 7 |  |
 |
 |
82(3g) |
| 8 | 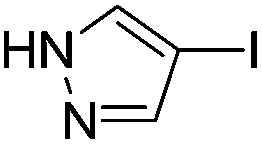 |
 |
 |
70(3h) |
| 9 |  |
 |
 |
82(3g)c |
| 10 | 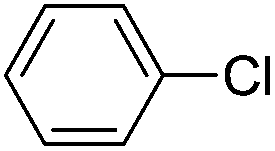 |
 |
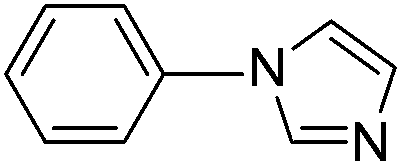 |
n.r.d |
| 11 |  |
 |
 |
70(3i) |
| 12 |  |
 |
 |
72(3j) |
| 13 |  |
 |
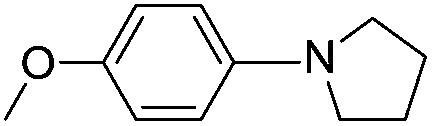 |
55(3k) |
| 14 | 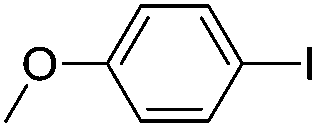 |
 |
 |
72(3l) |

To further expand the scope of this methodology, the C–S cross-coupling reaction, which is a powerful tool in organic synthesis,32 was studied under our optimal system (Table 3). Diphenyl disulfide was investigated as a representative example with aryl iodides having 4-NO2, 4-Cl, 2-Me, and 4-Ph substituents. The reactions occurred efficiently to afford the C–S cross-coupled products with up to 95% yield. In general, good yields were obtained for the cross-coupling of sterically unhindered aryl iodides. An ortho-substituted aryl iodide also gave satisfactory yields even with steric effects (Table 3, entry 5). No significant electronic effects were observed for either ortho- or para-substituted aryl iodides (entries 1–5). Under these conditions, the aryl iodide 4-iodo-1H-pyrazole showed moderate reactivity, leading to the cross-coupled product in 81% yield (Table 3, entry 6). Satisfactory yields were obtained in the coupling of bromobenzene with diphenyl disulfide (Table 3, entry 8). Interestingly, in all examples, only the unsymmetrical diaryl sulfide products were obtained in good yields under air. For the reaction mechanism, based on the results of experiments, which showed amino-containing D-glucosamine has better catalytic activity than other sugar-based ligands, we think the 1-OH and 2-NH2 of D-glucosamine may play an important role in coordination to CuI. The plausible mechanism for C–N and C–S couplings here is similar to our previously reported results.22b
|
|||
|---|---|---|---|
| Entry | Aryl halides | Product | Yieldb (%) |
| a Reaction conditions: CuI (0.1 mmol), D-glucosamine (0.1 mmol), 1 (1.0 mmol), 4a (0.6 mmol), Cs2CO3 (2.0 mmol), DMSO:H2O (3.0 mL 1:1), 100 °C under air, 12 h.b Isolated yield.c 2.0 mmol K2CO3 were used as base.d 24 h. |
|||
| 1 |  |
 |
95(92)(5a)c |
| 2 |  |
 |
82(5b) |
| 3 |  |
 |
85(5c) |
| 4 |  |
 |
80(5d) |
| 5 |  |
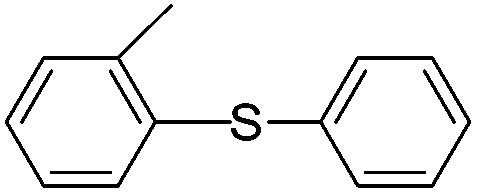 |
77(5e) |
| 6 |  |
 |
81(5f) |
| 7 |  |
 |
92(5g) |
| 8 |  |
 |
83(5g)d |

With this methodology in hand, we turned our attention to the synthesis of the clinically important anticancer drug nilotinib,31 a second-generation BCR-ABL tyrosine kinase inhibitor that shows greater efficacy than imatinib (Gleevec) in the treatment of chronic myelogenous leukemia (CML). Here, we apply our newly developed copper-catalyzed cross-coupling strategy to synthesize the key intermediate for the preparation of nilotinib. Key nilotinib intermediate 7 was prepared from aryl bromide 6, which was prepared from trifluoromethylbenzene in three steps in 62% yield (Scheme 2). When aryl bromide 6 (1.0 mmol) was treated with 4-methylimidazole (1.2 mmol) in the presence of 10 mol% CuI and 10 mol% D-glucosamine in DMSO–H2O at 100 °C for 10 h, nilotinib intermediate 7 was isolated in 81% yield as a single regioisomer.
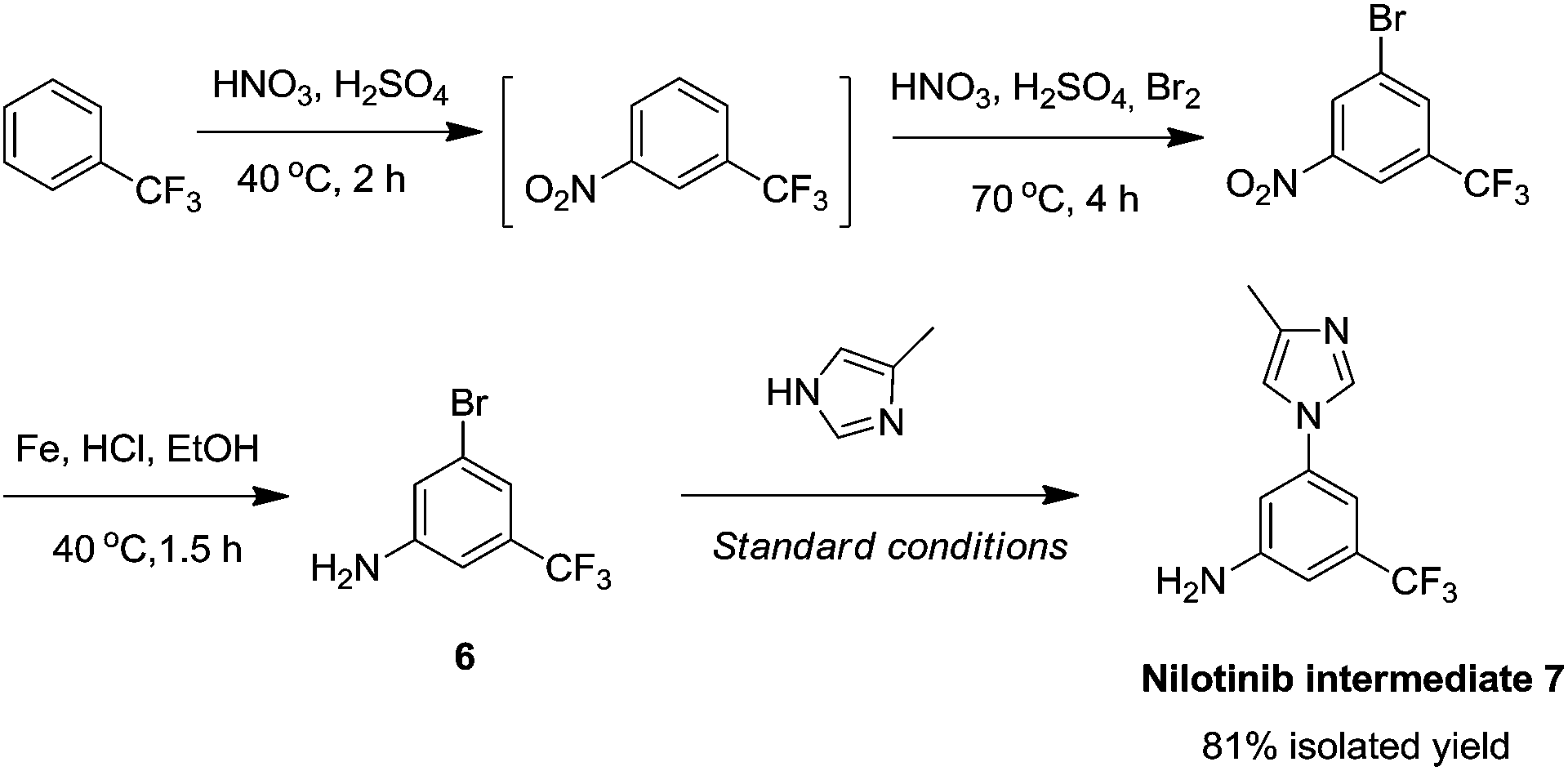 | ||
| Scheme 2 Application in synthesis of nilotinib intermediate 7. | ||
The incorporation of the cross-coupling strategy to the synthetic route provides a convenient and rapid method for chemical modifications to access a large range of structurally related analogues.
Conclusion
In summary, we have developed a simple, general, and efficient procedure for C–N and C–S bond formations via cross-coupling reactions of nitrogen and sulfur nucleophiles with both aryl iodides and bromides using D-glucosamine as a green ligand. Amides, amines, imidazoles, indole, pyrrolidine and diphenyl disulfide undergo reactions with aryl halides in the presence of 10 mol% D-glucosamine, 10 mol% CuI and 2 equiv. of Cs2CO3 in DMSO–H2O. The cross-coupling was successful with electron-rich and electron-poor aromatic iodides. Substrates bearing halides, free amino groups, trifluoromethyl and heterocycles were well tolerated. In general, this protocol not only provides a new catalytic system for the Buchwald-Hartwig amination reaction but also allows the synthesis of sulfur-containing compounds via more effective synthetic routes in high yields. The high water solubility of the ligand enables easy catalyst removal. In addition, the application of this catalytic system to the synthesis of nilotinib intermediate was also successfully demonstrated using commercially available substrates. Further investigation to broaden the scope of this catalytic system to other coupling reactions is currently ongoing in this laboratory.Experimental section
General information
The starting materials were commercially available and were used without further purification except solvents. The products were isolated by column chromatography on silica gel (200–300 mesh) using petroleum ether (60–90 °C) and ethyl acetate. Melting points were determined on an X-5 Data microscopic melting point apparatus. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer at ambient temperature with CDCl3 or DMSO-d6 as solvent unless otherwise noted and tetramethylsilane (TMS) as the internal standard. 1H NMR data were reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, m = multiplet and br = broad), coupling constant (J values, Hz). 13C NMR data were reported in terms of chemical shift (δ ppm). Mass spectra (EI-MS) were acquired on an Agilent 5975 spectrometer. Analytical thin layer chromatography (TLC) was performed on Merck precoated TLC (silica gel 60 F254) plates.General procedure for C–N cross-coupling reactions
A mixture of aryl halide (1 mmol), nitrogen nucleophile (1.2 mmol), CuI (0.1 mmol), D-glucosamine (0.1 mmol), and 3 mL of DMSO–H2O (1:1) in a tube was heated to 100 °C under air. The progress of the reaction was monitored by TLC using EtOAc and hexane as eluent. The cooled mixture was partitioned between ethyl acetate and water. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate twice. The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. After drying with anhydrous MgSO4 overnight, the liquid was analyzed by GC-MS and then the residue was purified on a short pad of silica gel using EtOAc and hexane as eluent. All compounds were characterized by 1H NMR and GC-MS spectroscopy, which were consistent with data reported in the literature.
Selected spectral data of the products
General procedure for C–S cross-coupling reactions
A mixture of aryl halide (1 mmol), phenyl disulfide (0.6 mmol), CuI (0.1 mmol), D-glucosamine (0.1 mmol), and 3 mL of DMSO–H2O (1:1) in a tube was heated to at 80–110 °C under air. Monitoring of the reaction, workup procedure, and purification of the C–S cross-coupled products were performed as described for the C–N cross-coupling reactions. All compounds were characterized by 1H NMR and GC-MS spectroscopy, which were consistent with those reported in the literature.
Acknowledgements
This work was supported by Science and Technology Plan of Zhejiang Province (no. 2014C31153), the National Natural Science Foundation of China (no. 21376058, 21302171) and Zhejiang Provincial Natural Science Foundation of China (No. LZ13B020001).Notes and references
- (a) K.-G. Liu, A. J. Robichaud, R. C. Bernotas, Y. Yan, J. R. Lo, M.-Y. Zhang, Z. A. Hughes, C. Huselton, G.-M. Zhang, J.-Y. Zhang, D. M. Kowal, D. L. Smith, L. E. Schechter and T. A. Comery, J. Med. Chem., 2010, 53, 7639 CrossRef CAS PubMed; (b) A. V. Ivachtchenko, E. S. Golovina, M. G. Kadieva, V. M. Kysil, O. D. Mitkin, S. E. Tkachenko and I. M. Okun, J. Med. Chem., 2011, 54, 8161 CrossRef CAS PubMed; (c) C. Shen, P.-F. Zhang, Q. Sun, S.-Q. Bai, T. S. Andy Hor and X.-G. Liu, Chem. Soc. Rev., 2014 10.1039/c4cs00239c.
- A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140–3152 RSC.
- Y. Liu and J. P. Wan, Org. Biomol. Chem., 2011, 9, 6873–6894 CAS.
- A. G. Ying, L. Liu, G. F. Wu, G. Chen, X. Z. Chen and W. D. Ye, Tetrahedron Lett., 2009, 50, 1653–1657 CrossRef CAS PubMed.
- C. Liu, H. Zhang, W. Shi and A. W. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed.
- C. Shen, H. J. Xia, H. Yan, X. Z. Chen, S. Ranjit, X. J. Xie, D. Tan, R. Lee, Y. M. Yang, B. G. Xing, K. W. Huang, P. F. Zhang and X. G. Liu, Chem. Sci., 2012, 3, 2388–2393 RSC.
- J. E. Andreasen, B. Shimpukade and T. Ulven, Green Chem., 2013, 15, 336–340 RSC.
- R. S. Xu, J. P. Wan, H. Mao and Y. J. Pan, J. Am. Chem. Soc., 2010, 132, 15531–15533 CrossRef CAS PubMed.
- (a) N. Zheng and S. L. Buchwald, Org. Lett., 2007, 9, 4749–4751 CrossRef CAS PubMed; (b) D. Maiti and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 17423–17429 CrossRef CAS PubMed; (c) L. B. Zhu, P. Guo, G. C. Li, J. B. Lan, R. G. Xie and J. S. You, J. Org. Chem., 2007, 72, 8535–8838 CrossRef CAS PubMed; (d) P. E. Maligres, S. W. Krska and P. G. Dormer, J. Org. Chem., 2012, 77, 7646–7651 CrossRef CAS PubMed.
- (a) J. H. Li, S. Benard, L. Neuville and J. P. Zhu, Org. Lett., 2012, 14, 5980–5983 CrossRef CAS PubMed; (b) J. M. Kallemeyn, Y. Y. Ku, M. M. Mulhern, R. Bishop and A. P. L. Jacob, Org. Process Res. Dev., 2014, 18, 191–197 CrossRef CAS; (c) M. A. Bhosale and B. M. Bhanage, RSC Adv., 2014, 4, 15122–15130 RSC.
- J. Bariwal and E. V. D. Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC.
- M. L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC.
- S. Venkatakrishnan, G. Veerappan, E. Elamparuthi and A. Veerappan, RSC Adv., 2014, 4, 15003–15006 RSC.
- P. Das and J. K. D. Brabander, Tetrahedron, 2013, 69, 7646–7652 CrossRef CAS PubMed.
- R. A. Altman and S. L. Buchwald, Org. Lett., 2006, 8, 2779–2782 CrossRef CAS PubMed.
- W. Zhu and D. W. Ma, J. Org. Chem., 2005, 70, 2696–2700 CrossRef CAS PubMed.
- P. E. Maligres, S. W. Krska and P. G. Dormer, J. Org. Chem., 2012, 77, 7646–7651 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13–31 RSC.
- D. P. Wang, F. X. Zhang, D. Z. Kuang, J. X. Yu and J. H. Li, Green Chem., 2012, 14, 1268–1271 RSC.
- K. G. Thakur, D. Ganapathy and G. Sekar, Chem. Commun., 2011, 47, 5076–5078 RSC.
- Y. Ou and N. Jiao, Chem. Commun., 2013, 49, 3473–3475 RSC.
- (a) C. Shen and P. F. Zhang, Curr. Org. Chem., 2013, 17, 1507–1524 CrossRef CAS; (b) M. Yang, H.-Y. Shen, Y.-Y. Li, C. Shen and P.-F. Zhang, RSC Adv., 2014, 4, 26295–26300 RSC.
- D. P. Cheng, F. F. Gan, W. X. Qian and W. L. Bao, Green Chem., 2008, 10, 171–173 RSC.
- C. Shen, H. X. Liao, F. Y. Shen and P. F. Zhang, Catal. Commun., 2013, 41, 106–109 CrossRef CAS PubMed.
- C. Shen, F. Y. Shen, H. J. Xia, P. F. Zhang and X. Z. Chen, Tetrahedron: Asymmetry, 2011, 22, 708–712 CrossRef CAS PubMed.
- C. Shen, F. Y. Shen, G. B. Zhou, H. J. Xia, X. Z. Chen, X. G. Liu and P. F. Zhang, Catal. Commun., 2012, 26, 6–10 CrossRef CAS PubMed.
- X. Ge, C. Qian, Y. B. Chen and X. Z. Chen, Tetrahedron: Asymmetry, 2014, 25, 596–601 CrossRef CAS PubMed.
- C. Shen, J. Xu, W. B. Yu and P. F. Zhang, Green Chem., 2014, 16, 3007–3012 RSC.
- M. Yang, H. Y. Shen, Y. Y. Li, C. Shen and P. F. Zhang, RSC Adv., 2014, 4, 26295–26300 RSC.
- S. Ueda and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 10364–10367 CrossRef CAS PubMed.
- (a) S. Ueda, M. J. Su and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 700–706 CrossRef CAS PubMed; (b) A. Stephan, A. Murat, E. Bernhard, K. Christoph, S. Joseph, M. Mark, P. Mahavir, S. W. Chung and X. Song, WO 20061356-40, 2006.
- (a) R. S. Schwab, D. Singh, E. E. Alberto, P. Piquini, O. E. D. Rodrigues and A. L. Braga, Catal. Sci. Technol., 2011, 1, 569–573 RSC; (b) A. R. Martin, D. J. Nelson, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Eur. J. Org. Chem., 2014, 3127–3131 CrossRef CAS; (c) L. Rout, T. K. Sen and T. Punniya-murthy, Angew. Chem., Int. Ed., 2007, 46, 5583–5586 CrossRef CAS PubMed; (d) Y.-Y. Liu, H. Wang, J. Zhang, J.-P. Wan and C.-P. Wen, RSC Adv., 2014, 4, 19472–19475 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, 13C NMR spectrum, GC/MS profile, HRMS profile. See DOI: 10.1039/c4ra11183d |
| This journal is © The Royal Society of Chemistry 2015 |