
Theoretical calculation based synthesis of a poly(p-phenylenediamine)–Fe3O4 composite: a magnetically recyclable photocatalyst with high selectivity for acid dyes†
Siwei Yang‡
*ab,
Caichao Ye‡ cd,
Xun Songa,
Lin Hea and
Fang Liao*a
cd,
Xun Songa,
Lin Hea and
Fang Liao*a
aChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637002, P. R. China. E-mail: liaozhang2003@163.com; Fax: +86 817 2568067; Tel: +86 817 2568067
bState Key Laboratory of Functional Materials for Informatics, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Science, Shanghai 20050, P. R. China. E-mail: yangsiwei@mail.sim.ac.cn; Fax: +86 021 62511070 ext. 420; Tel: +86 021 62511070 ext. 420
cSchool of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, P. R. China
dMaterials and Process Simulation Center, California Institute of Technology, Pasadena 91125, USA
First published on 17th October 2014
Abstract
A magnetically recyclable and highly photocatalytic poly(p-phenylenediamine)–Fe3O4 (PpPD–Fe3O4) composite photocatalyst was synthesised by a one-step chemical oxidation polymerisation based on theoretical calculation results. The band gap and selectivity of PpPD for acid dyes were studied by theoretical calculations. The calculation results showed that the PpPD molecules exhibited better conjugacy than PANI and that the band gap was more fit for use as a photocatalyst. The photocatalytic activities were evaluated by the degradation of various dyes. The stability and high selectivity of this composite was remarkable. The combination of Fe3O4 with PpPD lead to high photocatalytic activity in the degradation of the dyes under both ultraviolet, and visible light, irradiation, although Fe3O4 alone was inactive as a visible-light-driven photocatalyst.
1. Introduction
Nowadays, environmental pollution is a common problem that every country in the world faces. The amount of suitable water for drinking, agriculture, and farming, has declined through the years making this an important issue for waste water treatment and reduction.1 Synthetic dyes are used extensively in many industries such as: paper production, leather tanning, food technology, photoelectrochemical cells, and hair colourings.2,3 Synthetic dyes are a significant contributor to water pollution and have been proved to be harmful to humans.4–7 Semiconductor mediated photocatalysis has attracted increasing attention since water photolysis on a TiO2 electrode was discovered by Fujishima and Honda in 1972.8 Among various semiconducting materials, TiO2 is extensively investigated and widely used. On the other hand, a variety of inorganic photocatalysts such as: ZnO, CdS, SnO2, Ta3N5, and TaON are also used as photocatalysts.9–13 Unfortunately, due to their rapid recombination of charge carriers, very poor response to visible light, and critical drawback of photo-corrosion, many inorganic photocatalysts cannot be used under direct solar illumination, and their photoactivity and photostability are significantly reduced.14–16 Furthermore, many inorganic photocatalysts are expensive and hard to synthesise which limits their practical application.17As a new type of photocatalyst, conducting polymers (CP), such as polyaniline (PANI), poly-pyrrole, polythiophene, and their derivatives or composites have received increasing attention due to their advantages of low cost, ease of synthesis, high conductivity, and environmental stability.18–30 Wang et al.1 have synthesised a CuI/PANI super-hydrophobicity photocatalyst. Xiong et al.31 found a highly enhanced performance CoFe2O4–PANI magnetically recyclable photocatalyst. The combination of CoFe2O4 nanoparticles with PANI lead to a high photocatalytic activity in the degradation of dyes under visible light irradiation and the catalyst showed better catalytic activity for anionic dyes. However, how to improve the stability and catalytic activity of CP-based photocatalysts remains a challenge to researchers. Furthermore, the design of a CP-based photocatalyst which is easy to retrieve, or with high selectivity, is also a worthwhile research topic.
In this work, an Fe3O4–poly(p-phenylenediamine) (Fe3O4–PpPD) composite was prepared by an in situ polymerisation of the p-phenylenediamine (pPD) monomer in aqueous solution based on theoretical calculation results. All the theoretically calculated results were in good agreement with the experimental outcomes. Various dyes were used to evaluate the catalytic activity of the Fe3O4–PpPD composite. This composite can be separated easily by an external magnetic field and showed high selectivity and catalytic activity for acid dyes under both UV and visible light irradiation. The calculated results were consistent with the experiments. Fe3O4 which have no catalytic activity served as an electron sink and prevented the recombination of photoinduced electrons and holes. The PpPD is excited by UV or visible light and obtained photogenerated carriers and excited electrons, respectively. The excited state electrons in PpPD can readily migrate to the conduction band of Fe3O4. As PpPD is a good material for transporting holes, the photogenerated charges can migrate easily to the surface of the photocatalysts and photodegrade the adsorbed dye molecules.
2. Experimental section
2.1. Molecular design
Standard DFT was performed using the DMol3 (ref. 32) module. All geometries and energies were calculated using the generalised gradient approximation (GGA), using the functional PW91 (ref. 33–35) in combination with the double numerical polarised (DNP) basis set. All adsorption geometries were relaxed in three spatial directions, based on the geometrical optimisation criterion (an RMS force of 0.002 au Å−1 and an RMS displacement of 0.005 Å). The calculation of total energy and electronic structure was followed by the geometrical optimisation with an SCF tolerance of 1 × 10−5 au and a Fermi smearing of 0.005 Hartree. The best quality mesh size for numerical integration was then chosen. An unrestricted approach was applied such that the low-spin state, that is, a singlet for systems with an even-number of electrons and a doublet for systems with an odd-number of electrons was used. These low-spin states were, in general, energetically preferable.For the case of the adsorption configurations, the corresponding bond dissociation energy (EBDE) was calculated according to the expression
| EBDE = E(P–D) − (EP˙ + ED˙) | (1) |
2.2. Synthesis of magnetically recyclable PpPD–Fe3O4 photocatalyst and control sample
The PpPD–Fe3O4 magnetic composite was prepared as follows: 5.0 mL (30 mM) Fe(NO3)3 and 5.0 mL (10 mM) APS were mixed in a beaker. Then, 2.0 mL (1 M) HCl and 5.0 mL (10 mM) pPD were added to this solution. Shaking stopped after 0.5 min, and the reaction solution was kept for 24 h at 25 °C. The black product was collected by filtration, washed twice with distilled water and then dried in vacuum. The resulting precipitate was then dispersed in water and stored for characterisation and further use.The control sample (PpPD) was prepared as follows: 5.0 mL (10 mM) APS and 2.0 mL (1 M) HCl were mixed in a beaker. Then, 5.0 mL (10 mM) pPD were added to this solution. Shaking stopped after 0.5 min, and the reaction solution was kept for 24 h at 25 °C. The black product was collected by filtration, washed twice with distilled water and then dried in vacuum.
2.3. Characterizations
Powder X-ray diffraction (XRD) analysis was performed on a Bruker D8 Advanced diffractometer with Cu Kα radiation and a scanning angle 5° ≤ 2θ ≤ 80°. Scanning electron microscopy (SEM) measurements were made on a JSM-6510 (Rigaku, Japan) operated at an accelerating voltage of 20 kV. Infrared spectroscopy (FT-IR) was measured on a Bruker Vector-22 FT-IR spectrometer from 4000 to 500 cm−1 at room temperature. The Raman spectra were excited by radiation at 514.5 nm by a Jobin Yvon (France) LABRAM-HR Confocal Laser Micro-Raman Spectrometer.2.4. Photocatalytic activity measurement
Various dyes such as: bromocresol green (BG), bromocresol blue (BB), bromocresol purple (BP), Rhodamine b (RhB), neutral red (NR), methylene blue (MB), Sudan III (SIII), methyl orange (MO), and Congo red (CR), were used in this study. Before irradiation, the suspensions containing dyes and photocatalysts were magnetically stirred in the dark for 120 min to ensure the establishment of an adsorption/desorption equilibrium. Photo-irradiation was carried out using 500 W mercury and xenon lamps, respectively, as the light sources. The visible light source was equipped with a UV cutoff filters (JB420) to completely remove any radiation below 420 nm, thereby ensuring illumination by visible-light only. In all the photocatalytic degradation experiments, 25 mg of PpPD–Fe3O4 catalyst was added to 100 mL dye solution (50 mg L−1).2.5. Photocatalytic activity measurement
Adsorption experiments were conducted by adding 10 mg PpPD–Fe3O4 into the flask containing 100 mL of BB and MO solutions at an initial concentration of 25 mg L−1 under magnetic stirring for 2 h in the dark. Samples were collected after different time intervals (0, 1, 10, 20, 30, 60, 90, 120, 240, 360, and 720 min, respectively). The concentration of BB and MO was calibrated using the Beer–Lambert law at λmax values of 616 and 463 nm respectively by a Shimadzu UV-2550 UV-vis spectrophotometer.3. Results and discussion
3.1. Theoretical calculations
To design a new type of CP-based photocatalyst, the electric cloud distribution of PANI and PpPD were compared with the theoretical calculations. The molecular configuration and electric cloud distribution would help to evaluate the conjugation and carrier mobility of these polymers. To investigate the selectivity to acid and alkaline dyes, the band gap, bond lengths, and bond energy between PpPD and acid or alkaline dyes on different loci of PpPD were calculated.Previous reports confirmed that the PANI and PpPD were mixtures of oligomers, include dimer, trimer, and other oligomers.24–26,36,37 Considering this issue, four types of oligomers of PpPD were chosen: Fig. 1 shows all types of molecular structures used in the calculations, A1 was the molecular structure of the PANI, A2, A3, A4, and A5 were the molecular structures of PpPD. B1 to B4, and C1 to C4 were molecular models of PpPD, with which the BB combined by chemical adsorption. B1 to B4 were the BB combined with the N atoms (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) which were in the phenazine skeleton of PpPD. C1 to C4 were the BB combined with the N atoms (–NH2, NH) which were in the end-groups and residues of PpPD. D1 and D2 were the molecular models of PpPD, with which the MO combined at different sites (both the N atoms in the phenazine skeleton or end-groups) of PpPD.
N–) which were in the phenazine skeleton of PpPD. C1 to C4 were the BB combined with the N atoms (–NH2, NH) which were in the end-groups and residues of PpPD. D1 and D2 were the molecular models of PpPD, with which the MO combined at different sites (both the N atoms in the phenazine skeleton or end-groups) of PpPD.
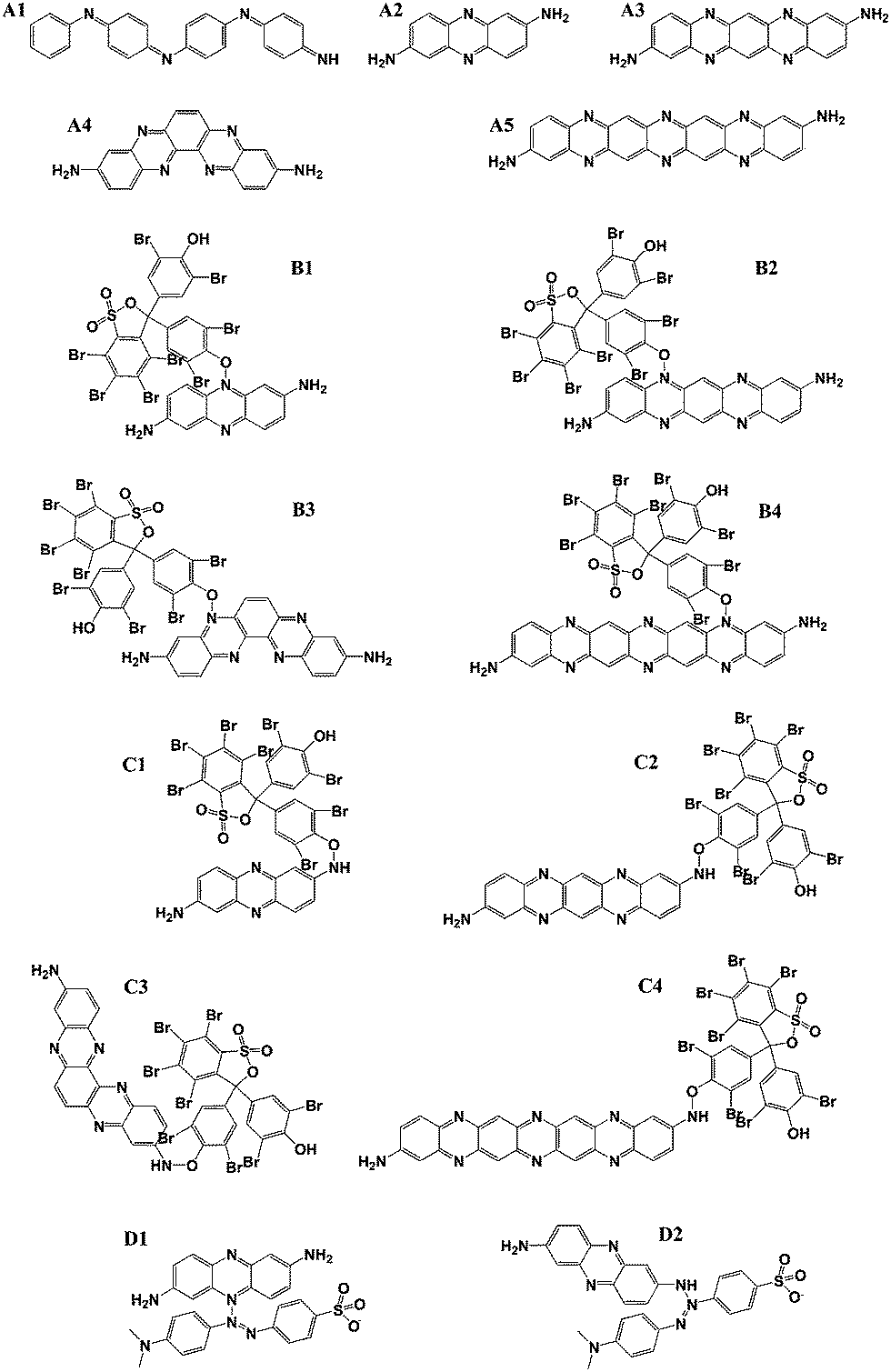 | ||
| Fig. 1 A schematic version of molecules in theoretical calculations. | ||
Fig. 2 shows the molecular configurations and electric cloud distribution of the HOMO–LUMO forms of PANI and PpPD, respectively. There was an angle between the planes of the adjacent benzene rings in PANI. However, all the atoms of the PpPD were mainly located in same plane, this suggested that the planarity of PpPD was better than PANI (Fig. 3). The phenazine skeleton25 limited the rotation of the benzene ring which improved the rigidity of PpPD. Additionally, the π electron cloud was delocalised over the whole molecular plane of the PpPD, while the π electron cloud only delocalised in the separation benzene rings of PANI. This indicated that PpPD molecules exhibited better conjugacy than PANI, and the carrier mobility of PpPD was also higher than that of PANI.
 | ||
| Fig. 2 Ball-and-stick model and theoretical electron distribution of the HOMO–LUMO energy states of PANI (A1) and PpPD (A2–5). | ||
 | ||
| Fig. 3 Schematic of the molecular structure of PANI and PpPD. | ||
The band gap (BG, eV) and the energy levels of molecular orbitals (HOMO, LUMO) are listed in Table 1. The BG of different types of PpPD polymer were 0.58 to 1.85 eV, this indicated that the PpPD acted as a type of semiconductor like PANI. Additionally the BG of PpPD was slightly lower than that of PANI,31 which showed that the PpPD might have well catalytic activity under visible light irradiation.
| Molecule | GB (eV) | HOMO (eV) | LUMO (eV) |
|---|---|---|---|
| A1 | 1.18745 | −5.08965 | −3.90220 |
| A2 | 1.85440 | −4.63269 | −2.77828 |
| A3 | 1.08584 | −4.68689 | −3.60105 |
| A4 | 2.05174 | −4.96839 | −2.91665 |
| A5 | 0.58921 | −4.68491 | −4.09570 |
Fig. 4 showed the molecular configuration of B1–B4, C1–C4, D1, and D2. The calculated results showed that both acid and alkaline dyes could be chemisorbed on PpPD. Table 2 showed the bond length (Å), and bond dissociation energy between PpPD and the dyes. The bond lengths were 1.4 to 1.5 Å when the BB or MO combined with the N atoms in both phenazine skeleton and end-groups of PpPD. This indicated that PpPD had strongly interacted with BB or MO. The short bond between BB and PpPD could be attributed to the strong interaction between acid groups (phenolic hydroxyl groups of BB) of acid dyes and alkalinity groups of PpPD. It was interesting that, although the bond lengths between dyes and PpPD were almost identical, the bond dissociation energy showed significant differences when the dyes combined with the N atoms at different loci of PpPD. The bond energy was lower than 100 kJ mol−1 when the dyes combined with the N atoms in the phenazine skeleton of PpPD.
 | ||
| Fig. 4 Ball-and-stick model of B1-4, C1-4, D1 and D2. | ||
| Molecule | Bond length (Å) | Bond dissociation energy (kJ mol−1) | BG (eV) |
|---|---|---|---|
| B1 | 1.418 | 10.0 | 1.44756 |
| B2 | 1.407 | 68.6 | 1.44521 |
| B3 | 1.442 | 6.30 | 1.42303 |
| B4 | 1.417 | 91.1 | 0.92779 |
| C1 | 1.441 | 179.8 | 1.67881 |
| C2 | 1.535 | 159.8 | 1.11635 |
| C3 | 1.447 | 201.0 | 2.05490 |
| C4 | 1.459 | 214.7 | 0.91419 |
| D1 | 1.414 | 24.5 | 0.37397 |
| D2 | 1.407 | 298.1 | 0.59422 |
Moreover, the bond energy increased (150 to 300 kJ mol−1) when the dyes combined with the N atoms in the end-groups and with the residues of PpPD. This change in bond energy could be attributed to the differences in N atoms and their loci on the PpPD. The large sterically hindrance of the N atoms in the phenazine skeleton mades the structure unstable and easy to desorbed when the dyes were adsorbed on PpPD. On the other hand, the amino and imino groups located in the end-groups and residues of PpPD, the steric effect was much weaker and the structure was more thermodynamically stable. There are many amino and imino groups in PpPD which ensured the high adsorption ability of the catalysts because of their low degree of polymerisation.31
All of the results demonstrated that the PpPD exhibited no selectivity for acid or alkaline dyes in the absorption progress. However, electrostatic repulsion might have affected the adsorption process when the alkaline dyes were closer to the N atoms of PpPD. Additionally, the selectivity in catalytic progression must be considered. The BGs of PpPD which adsorbed the dyes are also shown in Table 2. The BGs of PpPD changed after the dyes were adsorbed on the PpPD. The BG varied over a small range (0.9 to 2.0 eV) when the bromophenol blue combined with the N atoms in the phenazine skeleton or end-groups of the PpPD. Nevertheless, the BG decreased to 0.3 to 0.5 eV when the alkaline dye was adsorbed on the PpPD. The BG decreased: this may have resulted because the superoxide radical and hydroxyl free radical were harder to generate due to the low energy of the hole and electron. This may have caused the PpPD to degrade the alkaline dyes less readily.
Therefore, the PpPD might have selectivity to acid dyes in the photodegradation process. The alkaline dye could not easily remain in close proximity to the N atoms of PpPD due to the innate electrostatic repulsion in the reaction kinetics, although it was stable when the alkaline dye combined with the N atoms in both phenazine skeleton and the endgroups of PpPD. Additionally, the strong interaction between an alkaline dye and the PpPD caused the BG to decrease significantly which could result in a decrease in degradation ability towards alkaline dye. So the PpPD might be a selective photocatalyst in both adsorption and catalytic processes.
3.2. Characterization of PpPD–Fe3O4 photocatalyst
Fig. 5a showed the SEM imagines of the PpPD–Fe3O4 magnetic composite and control sample (PpPD); the PpPD–Fe3O4 was cuboidal. The surface of a single microcube showed no flaws, and the length of each side was approximately 500 nm while its length was approximately 1 to 2 μm. It was interesting that the microcubes formed into a globular superstructure and that the control samples were nanospheres. Uniform PpPD nanospheres with a mean diameter of 400 nm were seen in Fig. 2b. This showed that the Fe(NO3)3 was the main reason behind the morphology change in the PpPD–Fe3O4 magnetic composite. The changes of morphology of these products indicated that the Fe3+ was not only the dopant but also a template for the formation of the catalyst.23 | ||
| Fig. 5 SEM imagines of (a and b) PpPD–Fe3O4 magnetic composite and (c and d) PpPD. | ||
The Raman spectra were used to analyse the molecular structure of the PpPD–Fe3O4 composite (Fig. 6a). The peak at 1610 cm−1 was ascribed to the benzene ring. The peak at 1517 cm−1 corresponded to the N–H bending deformation mode and the bands at 1549 and 1594 cm−1 were ascribed to the C–C deformation of benzenoid rings and quinoid rings [35, 36], respectively. The spectra also revealed that the peaks located between 1361 and 1430 cm−1 were assigned to the C–N+ stretching modes of the delocalised polaronic charge carriers. The peak at 1164 cm−1 was due to the C–H inplane bending mode, which indicated the presence of a quinoid ring. The weak band at 1199 cm−1 was ascribed to the C–N stretching mode of polaronic units.21–24
 | ||
| Fig. 6 (a) Raman spectra of PpPD–Fe3O4, (b) XRD patterns of PpPD, PpPD–Fe3O4 and Fe3O4, (c) UV-vis spectra of PpPD–Fe3O4, (d) N2 adsorption desorption isotherms of PpPD–Fe3O4, the inset shows the pore size distributions of PpPD–Fe3O4. | ||
The FT-IR spectrum of PpPD–Fe3O4 composite is shown in Fig. S4, ESI.† The adsorption peaks at 3321, 3240, and 3210 cm−1 corresponded to the N–H stretching mode, and implied the presence of secondary amino groups. The strong peaks at 1601 and 1536 cm−1 were ascribed to CN and CC stretching vibrations in the phenazine structure, respectively. The peaks at 1239 and 1370 cm−1 were associated with C–N stretching in the benzenoid and quinoid imine units.37–40 The peaks at 900 and 830 cm−1 can be attributed to the out-of-plane deformation of C–H on a 1, 2, 4-tetrasubstituted benzene ring, which implied that the polymers had a basic phenazine skeleton. Moreover, the bands at 754 and 593 cm−1, characteristic of C–H out-of-plane bending vibrations of benzene nuclei in the phenazine skeleton, were also observed.41–44 The MALDI-TOF-MASS spectrum of this PpPD composite (Fig. S5, ESI†) confirmed that the PpPD was an oligomer and mainly composed of tripolymer, tetramer, and pentamer.
Fig. 6b showed the XRD patterns of the PpPD–Fe3O4 composite, pure Fe3O4, and PpPD. The two broad diffraction peaks at 20 and 30° were attributed to the amorphous structure of PpPD.37 Each diffraction peak of the Fe3O4 could be assigned to the cubic phase structure of Fe3O4 (JCPDS 65-3107). The peaks at 2θ values: 30, 35, 43, and 62.5°, could be indexed as the (220), (311), (400), and (422) crystal planes of cubic phase Fe3O4, respectively. There were no obvious diffraction peaks of PpPD in this composite which was similar to amorphous PANI in composites based on PANI with simple metal oxides.39
Fig. 6c showed the UV-vis spectra of the PpPD–Fe3O4 composite. The UV-vis absorption spectrum in H2O revealed absorption maxima at 545 and 350 nm, originating from the charge-transfer-excitation-like transition from the highest occupied energy level to the lowest unoccupied energy level and the π–π* transition, respectively.31,34 The results showed good agreement with the theoretical calculation and implied that the PpPD–Fe3O4 composite can be used as a UV or visible-light-driven photocatalyst. The N2 adsorption–desorption isotherm and corresponding pore size distribution of the PpPD–Fe3O4 composite are shown in Fig. 6d. This composite showed Type III isotherms, which exhibited almost no hysteresis loops. It indicated that N2 was hardly absorbed on the surface of the PpPD–Fe3O4 composite, and the phenomenon of autoacceleration was exhibited as the adsorption process occurred. The corresponding pore size distribution, as calculated by the BJH method, is shown in the insert to Fig. 6d: the pore size distribution of the PpPD–Fe3O4 composite consisted of small particle pores (2 to 12 nm), where the average pore diameter was 2 to 50 nm. The BET surface areas, total pore volumes, and average pore diameters of PpPD–Fe3O4 composite are summarised in Table. S1, ESI.† The decomposed XPS Fe 2p spectra of PpPD–Fe3O4 are shown in Fig. S7.†
3.3. Photodegradation of acid dyes with the magnetically recyclable PpPD–Fe3O4 photocatalyst
Various dyes were used in this study (Fig. 7). The photodegradation rates of BB on pure PpPD, Fe3O4, or PpPD–Fe3O4 under UV or visible light irradiation at 25 °C are shown in Fig. 8a and b, respectively: the Fe3O4 alone was a photocatalytically inactive compound under both UV and visible light. Moreover, the PpPD showed photocatalytic activity under both UV and visible light irradiation as was consistent with theoretical results. Additionally, the data showed that the combination of PpPD and Fe3O4 lead to a dramatic enhancement of photocatalytic activity under both UV and visible light irradiation. The BB was degraded completely in 24 h in the presence of PpPD–Fe3O4. | ||
| Fig. 7 The molecular structure dyes used in the experiment. | ||
 | ||
| Fig. 8 (a) Photodegradation of BB in the presence of PpPD–Fe3O4 under UV light irradiation, (b) photodegradation of BB in the presence of PpPD–Fe3O4 under visible light irradiation, the inset shows the picture of waste water containing BB before (left) and after (right) treatment by PpPD–Fe3O4 composite. | ||
To estimate the selectivity to acid dyes of PpPD–Fe3O4, Fig. 9a and b show the photocatalytic degradation of BG, RhB, MB, CR, and MO under UV and visible light in the presence of PpPD–Fe3O4. Fig 9c and d show the rate constants for various dyes (BG, BB, BP, RhB, NR, MB, SIII, MO, and CR). The photodegradation process could be expressed by pseudo-first-order kinetics:
 | (2) |
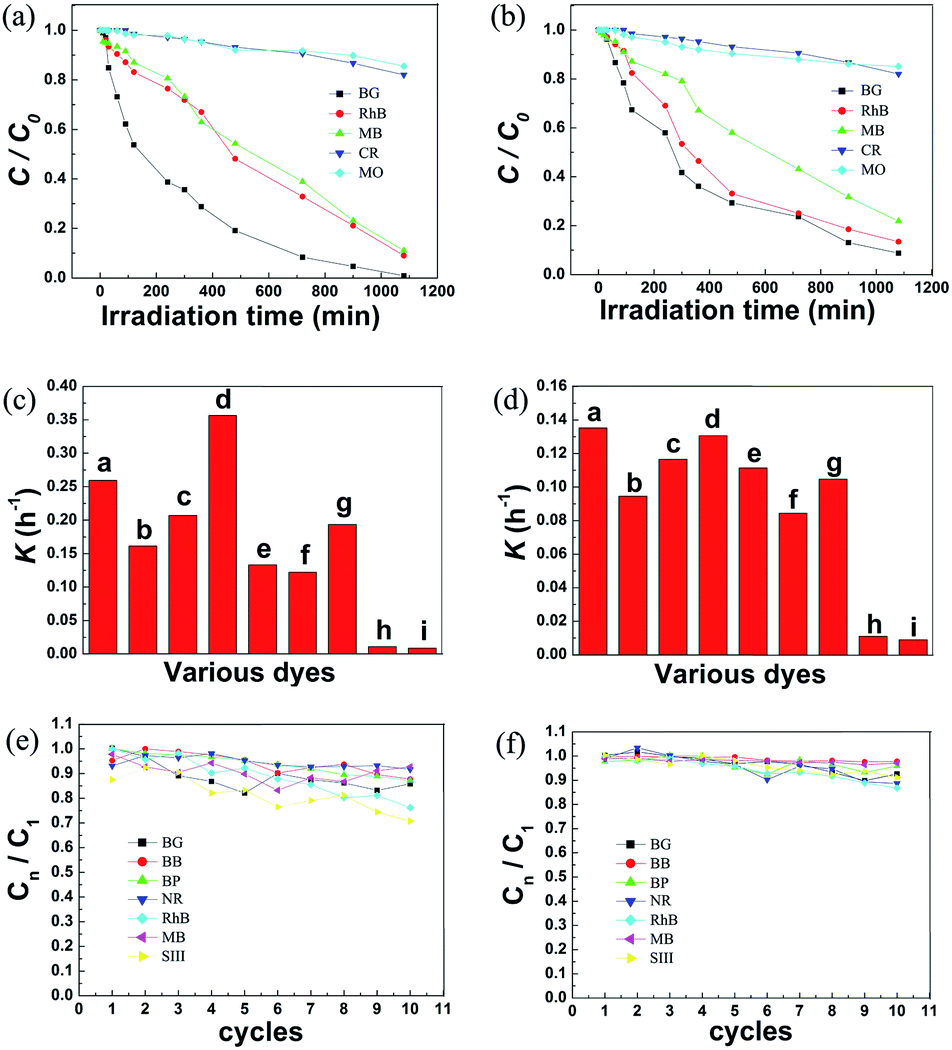 | ||
| Fig. 9 (a) Photodegradation of various dyes in the presence of PpPD–Fe3O4 under UV light irradiation, (b) photodegradation of various dyes in the presence of PpPD–Fe3O4 under visible light irradiation, (c) rate constants for various dyes under UV light irradiation, (d) rate constants for various dyes under visible light irradiation, (e) photodegradation of dyes in the presence of Fe3O4–PpPD under UV light irradiation in 10 cycles, the C1 is the concentration of BB at the end of the reaction (720 min) in the first cycle, the Cn is the concentration of BB at the end of the reaction (720 min) in the nst cycle, (f) photodegradation of dyes in the presence of Fe3O4–PpPD under visible light irradiation in 10 cycles. | ||
All the dyes degraded completely in 24 h in the presence of PpPD–Fe3O4 (Fig. S8–11, ESI†). This showed that the PpPD–Fe3O4 displayed good catalytic activities on the photodegradation of acid dyes (BG, BB, BP, RhB, NR, MB, and SIII) under both UV and visible light irradiation. Furthermore, the alkaline dyes (CR and MO) could not be degraded promptly by PpPD–Fe3O4. The photocatalytic degradations (Table 3) of the dyes were (in order): RB > BG > BP > SIII > BB > NB > MB ≫ CR > MO. The selectivity of the catalysts was significant and the results were in good agreement with the theoretical calculations.
| Dyes | UV light | Visible light | ||
|---|---|---|---|---|
| C/C0 | K (h−1) | C/C0 | K (h−1) | |
| BG | 0.00941 | 0.25922 | 0.08758 | 0.13529 |
| BB | 0.05472 | 0.16142 | 0.18233 | 0.09455 |
| BP | 0.02416 | 0.20684 | 0.12256 | 0.11662 |
| RB | 0.00164 | 0.35628 | 0.09521 | 0.13065 |
| NB | 0.09076 | 0.13331 | 0.13468 | 0.11138 |
| MB | 0.11076 | 0.12224 | 0.21893 | 0.08439 |
| SIII | 0.03076 | 0.19432 | 0.15173 | 0.10476 |
| MO | 0.82051 | 0.01099 | 0.82051 | 0.01099 |
| CR | 0.85557 | 0.00867 | 0.85116 | 0.00895 |
The photocatalytic activity of the recycled PpPD–Fe3O4 was also investigated, and the results are shown in Fig 9e and f. There was slight change in the photocatalytic activity of the catalyst in the first 10 cycles under visible or UV light irradiation. Further experimental data (Fig. S12 and S13, ESI†) verified the fact that the photocatalytic activity reduced significantly after 25 and 15 cycles under visible and UV light irradiation respectively. The lower stability of the catalyst could be attributed to the destructive effect of UV light on the PpPD. These results indicated that the PpPD–Fe3O4 catalyst showed good stability under both visible and UV light irradiation and was better than PANI.12–19 Furthermore the magnetism of the catalyst made it easy to recycle.
3.4. Possible mechanism of catalytic processes
To understand the photocatalytic mechanism behind this PpPD–Fe3O4 composite, the active oxidants generated in the photocatalytic process were trapped by disodium ethylenediaminetetraacetate dehydrate (EDTA-2Na) (a hole scavenger) and t-BuOH (a radical scavenger) to identify the main active oxidant in the photocatalytic reaction.12–17,31,45As shown in Fig. 10, both ultraviolet light and visible light irradiation were restrained, after the injection of EDTA-2Na, with regard to the photodegradation of BB. Futhermore, the photo-degradation ratio of BB was only slightly changed by the addition of t-BuOH under both ultraviolet light and visible light irradiation. This phenomenon suggested that holes were the main active species in the development and progress of photocatalysis.45 Furthermore, the photocatalytic activity was enhanced in the presence of Fe3O4. The separation efficiency of photoinduced electrons and holes was enhanced by the electronic interaction between Fe3O4 and PpPD.12–14
 | ||
| Fig. 10 (a) Plots of photogenerated carriers trapped in the system of photodegradation of BB under UV light by PpPD–Fe3O4, (b) plots of photogenerated carriers trapped in the system of photodegradation of BB under visible light by PpPD–Fe3O4. | ||
To further verify the selectivity of the catalyst, as predicted by theoretical calculation and confirmed by photocatalytic degradation experiment, the adsorption kinetics and isotherm of the BB on PpPD–Fe3O4 catalysts were studied. Kinetics experiments were carried out to verify whether or not the system followed the Langmuir–Hinshelwood mechanism. The equilibrium uptake could be calculated as follows:
 | (3) |
Fig. 11 shows the adsorption capacity of BB and MO over time in the presence of the PpPD–Fe3O4 composite: there was significant adsorption in the case of the BB. This was due to the acid groups of the BB which underwent chemical interactions with the alkalinity groups of PpPD. In contrast, the alkalinity dyes with alkalinity groups could not easily gain access to the alkalinity groups of PpPD because of electrostatic repulsion, eventually resulting in poor adsorption of the dyes. These experimentally measured results agreed with those calculated theoretically.
 | ||
| Fig. 11 (a) Plots of photogenerated carriers trapped in the system of photodegradation of BB under UV light by PpPD–Fe3O4, (b) plots of photogenerated carriers trapped in the system of photodegradation of BB under visible light by PpPD–Fe3O4. | ||
The rate kinetics of BB adsorption on the PpPD–Fe3O4 catalysts were analysed using the 1st-order rate kinetic model proposed by Lagergren (2),46 a pseudo-2nd-order model (3),47 and the rate equation as expressed by Weber and Morris (4):48
 | (4) |
 | (5) |
 | (6) |
Fig. 11b shows the Lagergren plot of log(qe − q) versus t (min) for adsorption of BB by the PpPD–Fe3O4 composite. The linearity of this plot indicated that a 1st-order mechanism did not follow the pseudo-1st-order kinetic model. The pseudo-2nd-order equation was expressed as a linear plot of (t/q) versus t (Fig. 11c) and was used to determine the rate constants and correlation coefficients. The pseudo-2nd-order rate constant (K′) and correlation coefficient (r2) for removal of BB from an aqueous solutions of PpPD–Fe3O4 composite were 0.00202 mg g−1 min−1 and 0.9991 respectively. This suggested that the values of correlation coefficients indicated a good fit for the pseudo-2nd-order model with the experimental data.49–51 Fig. 11d showed the Weber and Morris (intra-particle diffusion) plot for the adsorption of BB by PpPD–Fe3O4 composite: the plot was not linear over the entire time range. The kinetics experiments indicated that the mechanism of BB adsorption was complex and both the surface adsorption as well as intra-particle diffusion contributed to the rate determining step.48 This showed that the adsorption was mainly chemical adsorption51 and the calculation results agreed with the experimental measurements.
The BB adsorption isotherm data were correlated with the theoretical models of Langmuir52 and Freundlich:53
 | (7) |
 | (8) |
The linear plot of Ce/qe versus Ce and was used to determine the value of Langmuir constants qmax (mmol g−1) and b (L mg−1) (Fig. 11e). The Langmuir constants qmax and b were 505.05 mg g−1 and 9.68 × 103 L mg−1, respectively, furthermore, the correlation coefficient (r2) was 0.9973. The r2 values indicated a good fit to the Langmuir model for the experimental data.52,53 Fig. 11f shows the Freundlich plots for the adsorption of BB on PpPD–Fe3O4. The plot was not linear and this showed that the adsorption process did not follow the Freundlich adsorption model.53
The selectivity of PpPD–Fe3O4 in the photocatalytic degradation of acid dyes can be ascribed to the selective adsorption ability to acid dyes of the catalyst (Fig. 12). The interaction between the acidic groups of dyes and alkalinity groups of PpPD lead to the selective adsorption ability of PpPD–Fe3O4. The experimental results were in agreement with those calculated theoretically. Furthermore, the holes were the main active species in the photocatalytic progress. The enhancement of photoactivity could be attributed to the synergistic effect between Fe3O4 and PpPD. Fe3O4 which have no catalytic activity served as an electron sink and prevented the recombination of photoinduced electrons and holes. The PpPD is excited by UV or visible light and obtained photogenerated carriers and excited electrons, respectively. The excited state electrons in PpPD can readily migrate to the conduction band of Fe3O4. As PpPD is a good material for transporting holes,31,54,55 the photogenerated charges can migrate easily to the surface of the photocatalysts and photodegrade the adsorbed dye molecules.
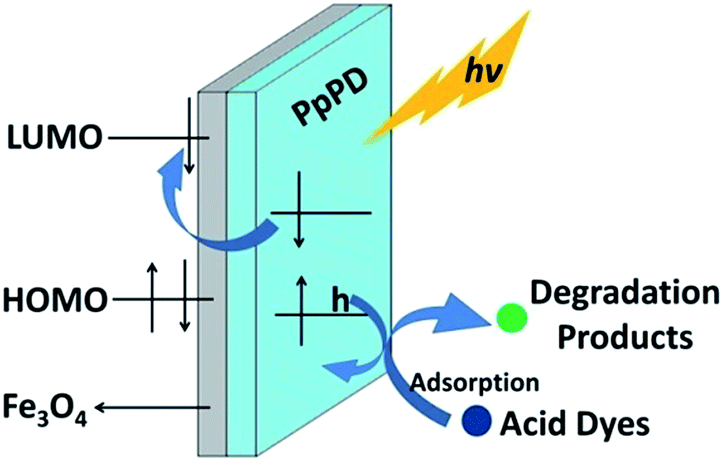 | ||
| Fig. 12 Schematic diagram illustrating the mechanism of adsorption and photodegradation over PpPD–Fe3O4. | ||
4. Conclusions
In summary, a magnetically recyclable and highly photocatalytic PpPD–Fe3O4 composite photocatalyst was synthesised by a one-step chemical oxidation polymerisation based on the results of theoretical calculations. The BG and selectivity of PpPD for the acid dyes were studied by theoretical calculation which showed that the PpPD molecules exhibited better conjugacy than PANI and that the BG was fit for use as a catalyst. PpPD displayed high selectivity in both adsorption and catalytic processes due to the electrostatic repulsion in its reaction kinetics: it significantly decreased the BG by a strong interaction between alkaline dye and PpPD. All the theoretically calculated results agreed with those obtained experimentally. Although Fe3O4 alone was an inactive visible-light-driven photocatalyst, the combination of Fe3O4 with PpPD lead to high photocatalytic activity for the degradation of the dyes under both ultraviolet and visible light irradiation.Acknowledgements
This work was supported by China West Normal University Fund (12B017) and China West Normal University Students Research Fund (42713070).Notes and references
- X. Wang, Y. Shen, A. Xie, L. Qiu, S. Li and Y. Wang, J. Mater. Chem., 2011, 21, 9641 RSC.
- Q. Z. Yu, M. Wang, H.-Z. Chen and Z.-W. Dai, Mater. Chem. Phys., 2011, 129, 666 CrossRef CAS PubMed.
- N. M. Mahmoodi, M. Arami, N. Y. Limaee and N. S. Tabrizi, Chem. Eng. J., 2005, 112, 191 CrossRef CAS PubMed.
- D. Yang, H. Liu, Z. Zheng, Y. Yuan, J.-C. Zhao, E. R. Waclawik, X. Ke and H. Zhu, J. Am. Chem. Soc., 2009, 131, 17885 CrossRef CAS PubMed.
- M. Zhu, P. Chen and M. Liu, ACS Nano, 2011, 5, 4529 CrossRef CAS PubMed.
- S. Xiong, Q. Wang and H. Xia, Synth. Met., 2004, 146, 37 CrossRef CAS PubMed.
- C. H. Kim, B.-H. Kim and K. S. Yang, Synth. Met., 2011, 161, 1068 CrossRef CAS PubMed.
- A. Fujishima, Nature, 1972, 238, 37 CrossRef CAS.
- Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei and J. Zhu, J. Phys. Chem. C, 2008, 112, 10773 CAS.
- R. Seoudi, M. Kamal, A. A. Shabaka, E. M. Abdelrazek and W. Eisa, Synth. Met., 2010, 160, 479 CrossRef CAS PubMed.
- M. Niu, F. Huang, L. Cui, P. Huang, Y. Yu and Y. Wang, ACS Nano, 2010, 4, 681 CrossRef CAS PubMed.
- H. Cui, K. Dwight, S. Soled and A. Wold, J. Solid State Chem., 1995, 115, 187 CrossRef CAS.
- R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Commun., 2005, 30, 3829 RSC.
- W. B. Lu, X. Y. Qin, H. Y. Li, A. M. Asiri, A. O. Al-Youbi and X. P. Sun, Part. Part. Syst. Charact., 2013, 30, 67 CrossRef CAS.
- J. Xu, S. Sun, Y. Cao, P. Lu, W. Li and K. Chen, Part. Part. Syst. Charact., 2014, 31, 459 CrossRef CAS.
- X. Qin, W. Lu, A. M. Asiri, A. O. Al-Youbi and X. Sun, Catal. Sci. Technol., 2013, 3, 1027 CAS.
- K. Saito, J. Kuwabara and T. Kanbara, Synth. Met., 2011, 16, 1150 CrossRef PubMed.
- R. Gangopadhyay and A. De, Chem. Mater., 2000, 12, 608 CrossRef CAS.
- J. Yan, T. Wei, B. Shao, Z. Fan, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 487 CrossRef CAS PubMed.
- J. Chen, L. Bai, M. Yang, H. Guo and Y. Xu, Synth. Met., 2014, 187, 108 CrossRef CAS PubMed.
- M. Omastová, K. Mosnáčková, P. Fedorko, M. Trchová and J. Stejskal, Synth. Met., 2013, 166, 57 CrossRef PubMed.
- B. H. Patil, A. D. Jagadale and C. D. Lokhande, Synth. Met., 2012, 162, 1400 CrossRef CAS PubMed.
- S. W. Yang, D. Liu, F. Liao, T. T. Guo, Z. P. Wu and T. T. Zhang, Synth. Met., 2012, 162, 2329 CrossRef CAS PubMed.
- S. W. Yang and F. Liao, Nano, 2011, 6, 597 CrossRef CAS.
- T. T. Zhang, S. W. Yang, J. Sun, X. B. Li, L. He, S. Yan, X. Y. Kang, C. S. Hu and F. Liao, Synth. Met., 2013, 181, 86 CrossRef CAS PubMed.
- S. W. Yang, S. Q. S. Huang, D. Liu and F. Liao, Synth. Met., 2012, 162, 2228 CrossRef CAS PubMed.
- S. W. Yang and F. Liao, Synth. Met., 2012, 162, 1343 CrossRef CAS PubMed.
- W. Lu, X. Qin, S. Liu, G. Chang, Y. Zhang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2012, 84, 5351 CrossRef CAS PubMed.
- X. Qin, S. Liu, W. Lu, H. Li, G. Chang, Y. Zhang, J. Tian, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Catal. Sci. Technol., 2012, 2, 711 CAS.
- Z. P. Wu, S. W. Yang, Z. Chen, T. T. Zhang, T. T. Guo, Z. F. Wang and F. Liao, Electrochim. Acta, 2013, 98, 104 CrossRef CAS PubMed.
- P. Xiong, Q. Chen, M. He, X. Sun and X. Wang, J. Mater. Chem., 2012, 22, 17485 RSC.
- B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- X. B. Li, S. W. Yang, J. Sun, P. He, X. G. Xu and G. Q. Ding, Carbon, 2014, 78, 38 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS.
- F. Liao, S. Yang, X. Li, L. Yang, Z. Xie, C. Hu, L. He, X. Kang, X. Song and T. Ren, Synth. Met., 2014, 189, 135 CrossRef CAS PubMed.
- F. Liao, S. Yang, X. Li, L. Yang, Z. Xie, C. Hu, S. Yan, T. Ren and Z. Liu, Synth. Met., 2014, 189, 126 CrossRef CAS PubMed.
- J. Tian, S. Liu and X. P. Sun, Langmuir, 2010, 26, 15112 CrossRef CAS PubMed.
- H. Q. Jiang, X. P. Sun, M. H. Huang, Y. L. Wang, D. Li and S. J. Dong, Langmuir, 2006, 22, 3358 CrossRef CAS PubMed.
- F. Liao, S. W. Yang, X. B. Li, S. Yan, C. S. Hu, L. He, X. Y. Kang, X. Song and T. Y. Ren, Synth. Met., 2014, 190, 79 CrossRef CAS PubMed.
- O. Akhavan, M. Choobatashani and E. Ghaderi, J. Phys. Chem. C, 2012, 116, 9653 CAS.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S.-T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430 CrossRef CAS PubMed.
- X. T. Chang, L. H. Dong, Y. S. Yin and S. B. Sun, RSC Adv., 2013, 3, 15005 RSC.
- X. B. Li, S. W. Yang, J. Sun, P. He, X. G. Xu and G. Q. Ding, Carbon, 2014, 78, 38 CrossRef CAS PubMed.
- S. Lagergren, K. Sven. Vetenskapsakad. Handl., 1898, 24, 1 Search PubMed.
- K. Periasamy, K. Srinivasan and P. R. Muruganan, J. Environ. Health, 1991, 33, 433 CAS.
- W. J. Weber and J. C. Morris, J. Sanit. Eng. Div., Am. Soc. Civ. Eng., 1963, 89, 31 Search PubMed.
- M. P. Candela, J. M. M. Martinez and R. T. Macia, Water Res., 1995, 29, 2174 CrossRef.
- M. Alibadi, K. Morshedzadeh and H. Soheyli, Int. J. Environ. Sci. Technol., 2006, 3, 321 CrossRef.
- M. A. Hossain, M. Kumita, Y. Michigami and S. Mori, J. Chem. Eng., 2005, 6, 402 CrossRef.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361 CrossRef CAS.
- K. Kadirvelu and C. Namasivayam, Adv. Environ. Res., 2003, 7, 471 CrossRef CAS.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953 CrossRef CAS PubMed.
- Z. Wang, R. Guo, G. Li, L. Ding, Y. Ou and Y. Tong, RSC Adv., 2011, 1, 48 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11138a |
| ‡ These authors are co-first authors. |
| This journal is © The Royal Society of Chemistry 2014 |