
MOF-derived microporous carbon as a better choice for Na-ion batteries than mesoporous CMK-3†
Abstract
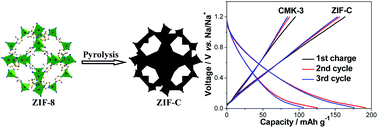
A metal–organic framework, 2-methylimidazole zinc salt (ZIF-8), is prepared through a scalable solution-precipitation method. Microporous carbon (ZIF-C) with a homogeneous pore size of 0.5 nm is obtained through direct pyrolysis of ZIF-8. Mesoporous carbon (CMK-3) with an average pore size of about 4.0 nm is achieved using SBA-15 as hard template. Microporous ZIF-C exhibits considerably higher capacity and better reversibility than mesoporous CMK-3 as anode material for Na-ion batteries. In addition, it is found that microporous ZIF-C can greatly reduce the irreversible capacity loss (reductive decomposition of electrolyte) of the first cycle and enhance the reversibility of Na+ storage during the subsequent cycles.
 Please wait while we load your content...
Please wait while we load your content...

