
Asymmetric allylation of sulfonyl imines catalyzed by in situ generated Cu(II) complexes of chiral amino alcohol based Schiff bases†
Debashis Ghoshab,
Prasanta Kumar Beraa,
Manish Kumarab,
Sayed H. R. Abdi*ab,
Noor-ul H. Khanab,
Rukhsana I. Kureshyab and
Hari C. Bajajab
aDiscipline of Inorganic Materials and Catalysis, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), G. B. Marg, Bhavnagar-364 002, Gujarat, India. E-mail: shrabdi@csmcri.org; Fax: +91 0278 2566970
bAcademy of Scientific and Innovative Research, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), G. B. Marg, Bhavnagar-364 002, Gujarat, India
First published on 24th October 2014
Abstract
A catalytic route for enantioselective synthesis of homoallyl amines through Cu(II)-Schiff base catalyzed reaction of allyltin with aryl, alkenyl-substituted N-sulfonylimines is described. The allylation reaction is promoted by a simple in situ generated Cu(II)-amino alcohol based Schiff base complex. The addition of allyltin to aldimines delivers the desired products up to 90% yield and 98% enantiomeric excess (ee). Based on experimental observations a probable mechanism was proposed for this reaction. The current methodology was extended to the synthesis of β-phenylalanine in good yield and very good enantioselectivity.
Introduction
Asymmetric allylation of carbonyl compounds has been well established1 and various efficient methods have been reported1,2 in this field. However, asymmetric allylation of imine,2c,3–8 particularly allylation of sulfonated imines6d–f,7a,b,8 is comparatively less explored. The allylation products i.e. homoallyl amines are imbedded within various biologically active compounds.9 Thus synthesis of enantio-enriched homoallyl amines is of current research interest. Transition metal catalyzed allylation of N-substituted imines3–7 is one of the most powerful tools for the preparation of chiral homoallyl amine. However, inexpensive copper catalyzed asymmetric allylation of imines have been rarely explored.7 Recently A. H. Hoveyda et al., reported (2011) enantioselective allylboration of substituted phosphinoylimines using N-heterocyclic carbine (NHC)–Cu complex.7d Although this efficient method gave excellent product yield and enantioselectivity, this method used extremely low temperature and required multistep synthesis of NHC-ligand. We thought of a simple in situ generated Cu(II)-chiral Schiff base type complex that can work under ambient reaction condition. Synthesis of the ligands follow one step condensation between a bisaldehyde and a chiral amino alcohol.10,11a The catalytic efficiency (with respect to both yield and enantioselectivity) is heavily influenced by the substitutions on amino alcohol part. Among various Schiff base ligands, tert-leucinol derived Schiff base ligand was found to be most suitable one in the above catalytic system. Besides this, presence of an additive in an appropriate amount has also influenced the enantioselectivity with a slight change in yield of allylation product. Involvement of the additive in the catalytic cycle was evidenced by UV-vis. as well as mass spectral studies. Our present investigation revealed that a simple chiral Schiff base-Cu(II) complex with a suitable additive is an efficient catalytic system for enantioselective allylation of sulfonyl imine.Results and discussion
Chiral Schiff base ligands (S,S)-1 to (S,R,S,R)-4 were prepared by the condensation of aromatic bis-aldehyde with different chiral amino alcohols (Scheme 1) by following literature procedure.10,11a,12 The ligand (S,S)-2′ was prepared by the reduction of (S,S)-2 with NaBH4 in MeOH (Scheme 1).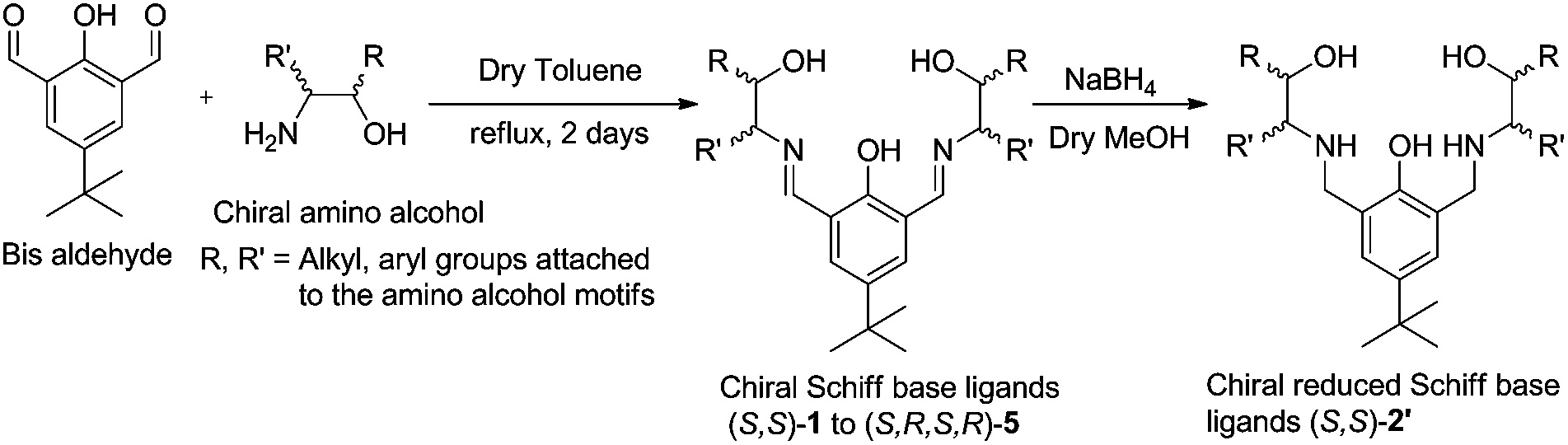 | ||
| Scheme 1 General synthesis of chiral ligands. | ||
The required substrates 1a-1p for the synthesis of enantiorich homoallyl sulfonamines 2a-2p can easily be prepared in a single step by the condensation of corresponding aldehydes with cyanuric chloride in good yields.11a We began our investigation by using N-(4-chlorobenzylidene)-4-methylbenzene sulfonamide (1b) as a representative substrate, allyltributyltin as an allylating agent and in situ generated chiral Schiff base-Cu(II) complexes of ligands (S,S)-1–3 and (S,R,S,R)-4 as catalysts in CH2Cl2 at RT. The results are summarized in Table 1.
|
|||
|---|---|---|---|
| Entry | Ligands | Yieldb (%) | eec (%) |
| a All the reactions were carried out by using substrate N-(4-chlorobenzylidene)-4-methylbenzene sulfonamide (0.3 mmol), allyltributyltin (0.45 mmol), and catalyst (10 mol%) in CH2Cl2 at RT.b Isolated yields after column chromatography.c ee determined by chiral HPLC using Daicel Chiralcel OD-H column. | |||
| 1 | (S,S)-1 | 40 | 30 |
| 2 | (S,S)-2 | 60 | 64 |
| 3 | (S,S)-2′ | 30 | 10 |
| 4 | (S,S)-3 | 25 | 20 |
| 5 | (S,R,S,R)-4 | 10 | 12 |

At first we have checked the efficiency of in situ generated L-valinol derived ((S,S)-1) Schiff base-Cu(II) complex for the above allylation reaction, where homoallyl sulfonamine was obtained in moderate yield (40%) and low enantioselectivity (ee, 30%) (Table 1, entry 1). Then we have altered the amino alcohol part in the Schiff base ligand by changing L-valinol with L-tert-leucinol ((S,S)-2) (Fig. 1). This modification resulted in a dramatic improvement of product yield (60%) and ee (64%) (entry 2). To get better yield as well as enantioselectivity, we have varied the structural unit of amino alcohol part as in ligands (S,S)-3 and (S,R,S,R)-4, but the results obtained with (S,S)-2 were better than other ligands (entries 2, 4 and 5) used in the present study. We have also checked the efficiency of the reduced Schiff base ligand (S,S)-2′, but we got the product in poor yield and ee with this system (entry 3).
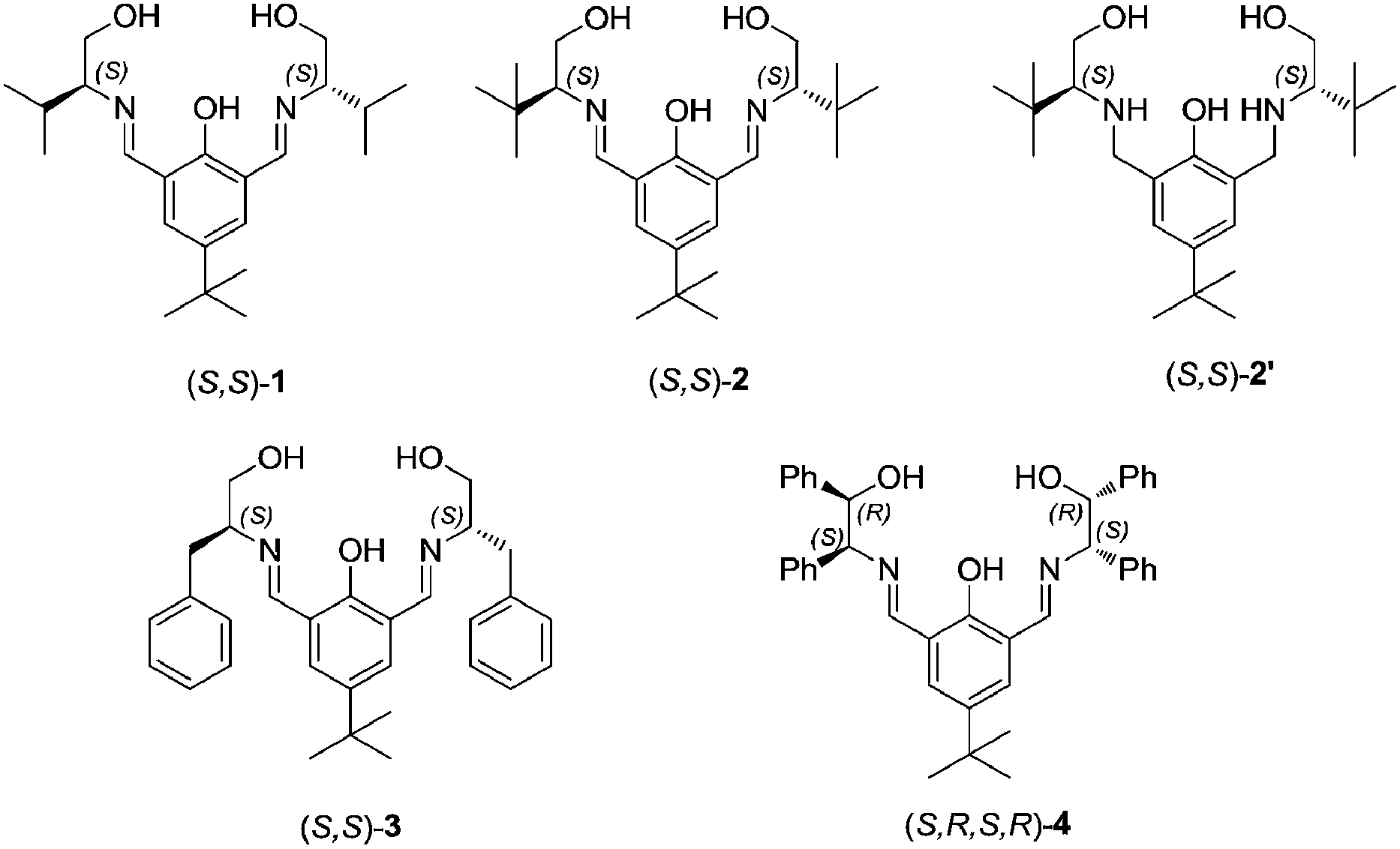 | ||
| Fig. 1 Chiral ligands used in the present study. | ||
A good level of enantioselectivity was already achieved in absence of an additive (Table 2, entry 1) but addition of an additive (10 mol%) among L-tert-leucine, L-valinol and L-tert-leucinol greatly influenced ee of the product (entries 2–4). Among these L-tert-leucinol (10 mol%) had a very positive influence on the allylation reaction. The allylation results were further improved by increasing the amount of L-tert-leucinol to 20 mol% (entry 5), but a further increase in its amount (30 mol%) was of little use (entry 6). It is worth mentioning here that L-tert-leucinol itself can act as ligand, therefore we used it as ligand and conducted the allylation reaction in the absence of (S,S)-2 keeping other parameters constant. However, yield as well as ee of the allylation product were found to be poor (entry 7) implying that a combination of L-tert-leucinol and (S,S)-2 with copper is forming a highly active and enantioselective catalyst. Furthermore, it is known that different copper salts have variable geometry and reactivity, hence we varied copper sources viz., Cu(OAc)2·H2O, CuOTf, Cu(OTf)2 and Cu(acac)2 (entries 5, 9–11) and found that Cu(OAc)2·H2O was more efficient (entry 5). The optimum reaction must be carried out in presence of 4 Å molecular sieves, in absence of which a large decreased in product yield as well as ee (entry 8) was observed due to the moisture sensitive nature of sulfonyl imine.
|
|||||
|---|---|---|---|---|---|
| Entry | CuLn | Additive | Additive amount (mol%) | Yieldb (%) | eec (%) |
| a As per Table 1.b Isolated yield after column chromatography.c ee determined by chiral HPLC using Daicel Chiralcel OD-H column.d Reaction was done in the absence of ligand (S,S)-2.e Reaction was carried out in absence of 4 Å MS. | |||||
| 1 | Cu(OAc)2·H2O | — | — | 60 | 64 |
| 2 | Cu(OAc)2·H2O | L-tert-Leucine | 10 | 45 | 20 |
| 3 | Cu(OAc)2·H2O | L-Valinol | 10 | 56 | 70 |
| 4 | Cu(OAc)2·H2O | L-tert-Leucinol | 10 | 65 | 86 |
| 5 | Cu(OAc)2·H2O | L-tert-Leucinol | 20 | 66 | 94 |
| 6 | Cu(OAc)2·H2O | L-tert-Leucinol | 30 | 65 | 94 |
| 7 | Cu(OAc)2·H2Od | L-tert-Leucinol | 20 | 20 | 16 |
| 8 | Cu(OAc)2·H2Oe | L-tert-Leucinol | 20 | 30 | 60 |
| 9 | CuOTf·toluene | L-tert-Leucinol | 20 | 35 | 24 |
| 10 | Cu(OTf)2 | L-tert-Leucinol | 20 | 50 | 40 |
| 11 | Cu(acac)2 | L-tert-Leucinol | 20 | 20 | 16 |

The effect of solvent and catalyst loading were evaluated under so far optimized reaction condition (Table 2, entry 5) and the data are summarized in Table 3. Catalytic runs conducted in THF, CH3CN, CHCl3 or in toluene were not as effective as those in CH2Cl2 (entries 2, 6–10). Further, catalyst loading of 10 mol%, was found to be optimum under the above reaction condition (entries 1–3). Additionally, it appeared that 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.6 metal/ligand ratio is the best to achieve highest enantioselectivity (entries 2, 4 and 5).
:0.6 metal/ligand ratio is the best to achieve highest enantioselectivity (entries 2, 4 and 5).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. loading (mol%) | Cu(OAc)2·H2O/(S,S)-2 | Solvent | Time (h) | Yieldb (%) | eec (%) |
| a As per Table 1.b Isolated yield after column chromatography.c ee determined by chiral HPLC using Daicel Chiralcel OD-H column. | ||||||
| 1 | 5 | 1:0.6 |
CH2Cl2 | 65 | 50 | 76 |
| 2 | 10 | 1:0.6 |
CH2Cl2 | 46 | 66 | 94 |
| 3 | 15 | 1:0.6 |
CH2Cl2 | 45 | 69 | 88 |
| 4 | 10 | 1:1 |
CH2Cl2 | 46 | 67 | 90 |
| 5 | 10 | 1:1.5 |
CH2Cl2 | 46 | 66 | 90 |
| 6 | 10 | 1:0.6 |
CHCl3 | 60 | 50 | 60 |
| 7 | 10 | 1:0.6 |
THF | 72 | 35 | 40 |
| 8 | 10 | 1:0.6 |
CH3CN | 85 | 34 | 46 |
| 9 | 10 | 1:0.6 |
Toluene | 90 | 20 | 30 |

We further examined the chiral Schiff base-Cu(II) complex-promoted allylation reaction of 1b with other allylating agents (Table 4). Tetra-allyltin showed activity similar to the allyltributyltin but with significantly lower enantioselectivity (entries 2 and 4). In a marked contrast the reaction using allylsilanes did not proceed at all (entries 4 and 5). When we used (pinacolato)allylboron as an allylating agent we got homoallyl amine in good yield with very low enantioselectivity (entry 7). After fixing the allylating agent we optimized its amount and the results showed its 1.5 equivalent (with respect to 1b) was sufficient for getting highest product yield and enantioselectivity (entry 2).








The optimal condition established for the enantioselective allylation of N-(4-chlorobenzylidene)-4-methylbenzene sulfonamide 1b (Table 4, entry 2) was applied to other aromatic as well as α,β-unsaturated sulfonyl imines.
The desired homoallyl sulfonamines 2a-2p were obtained in mostly good yield with high enantioselectivities ranging from 98 to 66% enantiomeric excess (Table 5). Para- and meta-substituted aromatic imines gave good yield (except electron donating e.g. p-methoxy group) and excellent enantioselectivity (entries 2–10) as compared to their ortho-counterpart (entries 11 and 12). The present catalytic system works very well for bulkier aromatic imines (like 1-naphthyl and 2-naphthyl imines; entries 13 and 14) as well as α,β-unsaturated sulfonyl imines (e.g., trans-cinnamyl and alpha-methyl-trans-cinnamyl imines; entries 15 and 16).
|
||||
|---|---|---|---|---|
| Entry | Substrate | R1/R2 | Yieldb (%) | eec (%) |
| a All the reactions were carried out by using substrates 1a-1o (0.3 mmol), allyltributyltin (0.45 mmol), and catalyst (10 mol%) in CH2Cl2 at RT.b Isolated yields after column chromatography.c ee determined by chiral HPLC using OD-H, AD-H, IA chiral column. | ||||
| 1 | Ph | C6H5/Me (1a) | 2a (60) | 80 |
| 2 | p-ClC6H4 | p-ClC6H4/Me (1b) | 2b (66) | 94 |
| 3 | p-ClC6H4 | p-ClC6H4/H (1c) | 2c (58) | 90 |
| 4 | p-ClC6H4 | p-ClC6H4/NO2 (1d) | 2d (68) | 78 |
| 5 | p-BrC6H4 | p-BrC6H4/H (1e) | 2e (60) | 98 |
| 6 | p-FC6H4 | p-FC6H4/Me (1f) | 2f (69) | 90 |
| 7 | p-NO2C6H4 | p-NO2C6H4/Me (1g) | 2g (67) | 80 |
| 8 | p-CF3C6H4 | p-CF3C6H4/Me (1h) | 2h (65) | 76 |
| 9 | p-MeOC6H4 | p-MeOC6H4/H (1i) | 2i (30) | 84 |
| 10 | m-ClC6H4 | m-ClC6H4/Me (1j) | 2j (64) | 82 |
| 11 | o-ClC6H4 | o-ClC6H4/Me (1k) | 2k (70) | 66 |
| 12 | o-FC6H4 | o-FC6H4/Me (1l) | 2l (65) | 78 |
| 13 | 1-Naphthyl | 1-Naphthyl/Me (1m) | 2m (90) | 92 |
| 14 | 2-Naphthyl | 2-Naphthyl/Me (1n) | 2n (84) | 94 |
| 15 | trans-Cinnamyl | (E)-C6H5CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH/Me (1o) CH/Me (1o) |
2o (76) | 86 |
| 16 | Alpha-methyl-trans-cinnamyl | (E)-C6H5CHC(CH3)/Me (1p) |
2p (75) | 94 |

The present asymmetric allylation protocol was successfully extended to the synthesis of enantioenriched β-amino acid 7 (Scheme 2). β-Amino acids13 are important motifs which serve as precursors to β-lactams, constituents of several medicinally important compounds,14 and most importantly as monomers in the synthesis of peptidomimetic β-peptides.15 Compound 2a was oxidized with NaIO4 in the presence of a catalytic amount of OsO4 to form the corresponding aldehyde, which without purification was further oxidized to furnish the desired tosyl-protected β-amino acid 7 with an overall yield of 60% with retention of optical purity.
 | ||
| Scheme 2 Synthesis of enantioriched β-amino acid 7. | ||
Mechanism
To support the mechanism as given in Scheme 3 for the catalytic allylation reaction, a stepwise UV-vis. spectral study was carried out with N-(4-chlorobenzylidene)-4-methylbenzene sulfonamide (1b) and allyltributyltin in CHCl3 as solvent at RT (Fig. 2A–C). | ||
| Scheme 3 Proposed catalytic cycle. | ||
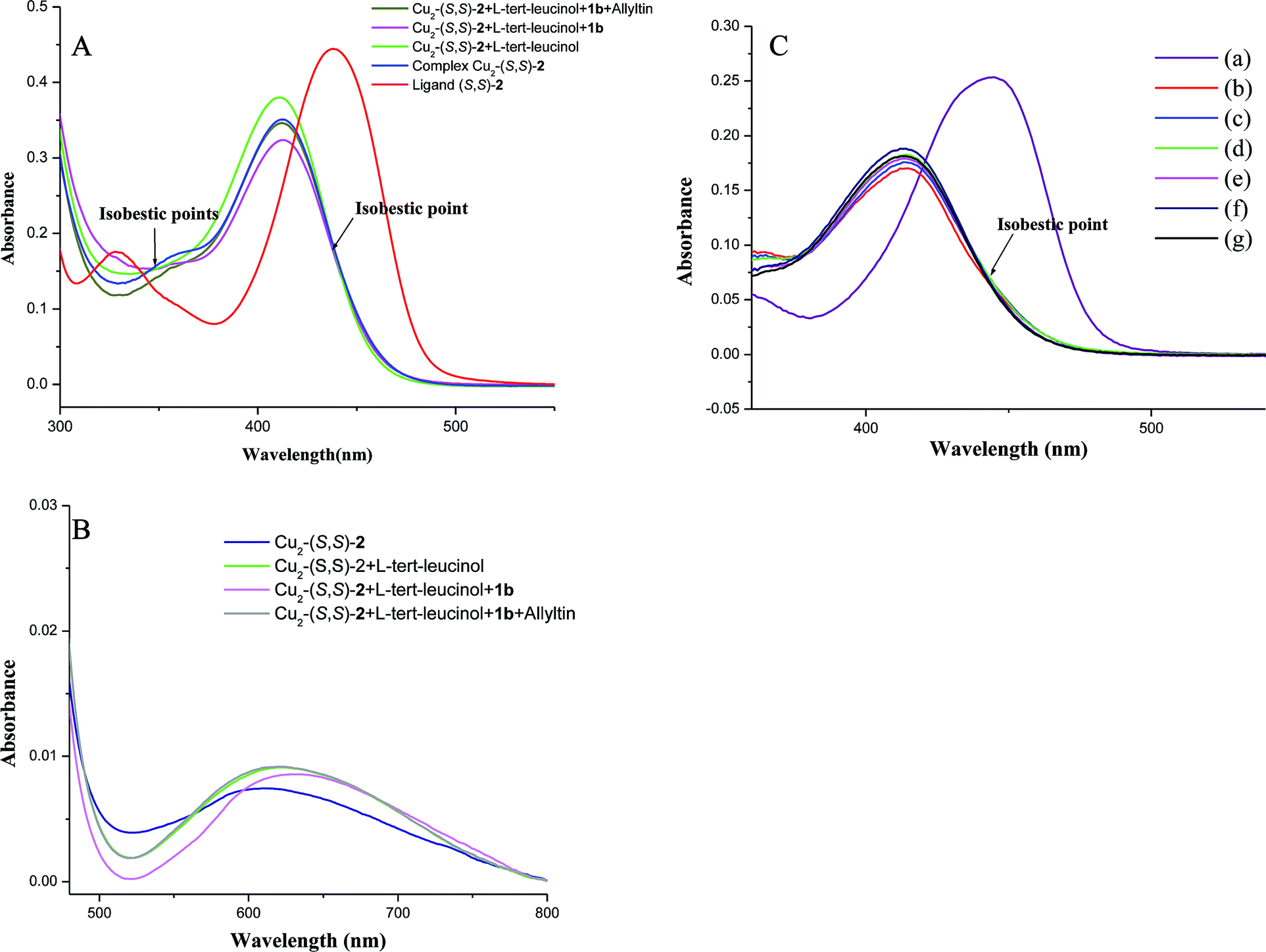 | ||
| Fig. 2 (A) UV-vis spectra of ligand, in situ generate complex and reaction mixture after sequential addition of additive, substrate and allyltributyltin. (B) UV-vis spectra (for d–d transition) of in situ generate complex and reaction mixture after sequential addition of additive, substrate and allyltributyltin. (C) UV-vis spectra of ligand, in situ generate complex and the reaction mixture with varying concentration of tert-leucinol: (a) ligand; (b) in situ generate complex; (c) complex:additive (1:0.2); (d) complex:additive (1:0.4); (e) complex:additive (1:0.6); (f) complex:additive (1:0.8); (g) complex:additive (1:1). | ||
Initially, in situ formation of the complex was confirmed by UV-vis (strong blue shift i.e. from 438 nm to 412 nm, Fig. 2A) as well as ESI-MS spectral analysis (Fig. 3). After the addition of additive (L-tert-leucinol) to the complex solution, the intensity of LMCT band at ∼410 nm has increase significantly whereas d–d band was red shifted (from 608 nm to 621 nm) with isobestic points at ∼348 nm and ∼438 nm suggests the formation of intermediate I-1 (Scheme 3).
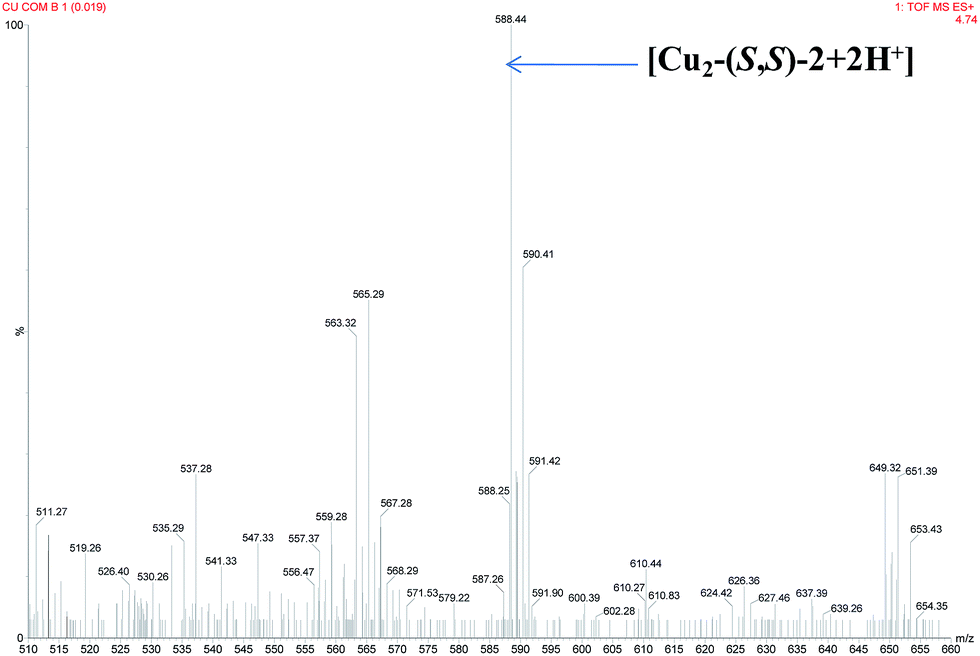 | ||
| Fig. 3 ESI-MS spectrum of the in situ generated complex was taken in methanol. | ||
To further confirm this interaction additional experiment on varying the amount of tert-leucinol to the in situ generated copper complex was carried out and was monitored on UV-vis and the spectra (Fig. 2C) that clearly show isobestic point at ∼441 nm, and thereby additionally supports the formation intermediate I-1 during the catalytic cycle.
This intermediate I-1 was further confirmed by ESI-MS spectral analysis where new molecular mass peak equivalent to [I-1 + H+] species (Fig. 4) is clearly visible. After the addition of substrate to the above solution, LMCT band maxima slightly diminished with isobestic points at ∼348 nm and ∼438 nm. A significant red shift in the d–d band (from 621 nm to 631 nm) confirms the direct co-ordination of the substrate to the vacant d orbital of the copper through the lone pair of the nitrogen atom of the N-tosylimine. The generation of the isobestic points may be due to the change in geometry of the copper complex and this can be attributed to the attachment of the L-tert-leucinol and the substrate to the complex to form an intermediate I-2 (Scheme 3).
 | ||
| Fig. 4 ESI-MS spectrum of the in situ generated complex after interaction with additive was recorded in methanol. | ||
The intermediate I-2 was further confirmed by ESI-MS spectral analysis where new molecular mass peak corresponding to [I-2 + CH3OH + H+] species (see Fig. 1 in ESI†). On further addition of allyltributyltin to the reaction mixture there was no observable change in the spectrum. The addition of allyltributyltin follows product formation and regenerates the intermediate I-1 for the next cycle.
Conclusion
In conclusion, a new copper based catalytic protocol was developed for asymmetric allylation reaction of aryl, alkenyl-substituted N-sulfonylimines using allyltributyltin as allylating agent. More importantly, the present catalytic system is simple and works under ordinary reaction condition giving high yields (up to 90%) of the homoallyl amines with excellent enantioselectivities (ee up to 98%) as compared to the other reports. Additionally, the current methodology was extended to the synthesis of β-phenylalanine in good and very good enantioselectivity.Experimental section
Different aldehydes and reagents were used as received. The imine substrates (1a-1o) were prepared following literature procedure.11a All the solvents were dried using standard procedures,16 distilled and stored under activated molecular sieves. NMR spectra were obtained with a Bruker F113V spectrometer (200 and 500 MHz) and are referenced internally with tetra-methylsilane (TMS). Splitting patterns were reported as s, singlet; d, doublet; dd, doublet of doublet; t, triplet; q, quartet; m, multiplet; br, broad. Enantiomeric ratio (er) values were determined by HPLC (Shimadzu SCL-10AVP) using Daicel Chirl-pak OD-H, AS-H, IA, IB and IC chiral columns with 2-propanol/hexane as eluent. For the product purification flash chromatography was performed using silica gel 100–200 mesh.General procedure for preparation of catalysts (S,S)-1 to (S,R,S,R)-411a
To a solution of 4-tert-butyl-2,6-diformylphenol (300 mg; 1.45 mmol) in dry toluene chiral (S)-tert-leucinol (341 mg; 2.91 mmol) was added and the reaction was vigorously stirred for 2 days under reflux condition. In two days yellow coloured precipitate of desired product was formed in the reaction mixture which was filtered, washed twice with cold methanol and dried to get desired ligand (S,S)-2 in sufficient purity (yield, 515 mg; 88%) and was fully characterised by NMR and elemental analysis before its use in catalytic reaction. All other Schiff base ligands (S,S)-1 to (S,R,S,R)-4 were prepared following the above reaction procedure.Typical procedure for preparation of ligand (S,S)-2′
To the solution of (S,S)-2 (405 mg, 1 mmol) in 20 mL dry methanol NaBH4 (303 mg, 4 × 2 mmol) was added portion wise in four equal parts. The reaction was monitored by TLC. After completion of the reaction (10 h), solvent was completely removed under reduced pressure. Then the reaction mass was washed by water and extracted with CH2Cl2 and dried by Na2SO4. Further purification was done by flash column chromatography (EtOAc/hexane = 1:4) using silica gel (100–200 mesh). White solid. yield: 90%; m. p.: 150–152 °C. 1H NMR (CDCl3, 500 MHz): δ = 0.91 (s, 18H), 1.19 (s, 9H), 2.35 (m, 2H), 3.52–3.78 (m, 4H), 3.89–4.03 (m, 4H), 4.67 (br, 4H), 6.97 (s, 2H). 13C NMR (CDCl3, 125 MHz): δ = 27.24, 29.63, 31.52, 33.99, 52.14, 60.59, 67.65, 123.39, 125.58, 141.46, 154.42. TOF-MS (ESI+) calcd [M + H+] for (C24H44N2O3) 409.34, found: 409.66.
General procedure for Cu(II)-Schiff base complex catalyzed asymmetric allylation reaction of tosylimines 1a-1o
To a mixture of Cu(OAc)2·H2O (0.03 mmol), ligand (S,S-2) (0.018 mmol) and 60 mg of powdered activated 4 Å MS in a nitrogen-filled 5 mL reactor, dry DCM (2 mL) was added. After the mixture was stirred at 27 °C for 2 h, L-tert-leucinol (0.06 mmol) was added and again stirred for 30 minutes. To it sulfonimines 1a-1o (0.3 mmol) followed by allyltributyltin (0.45 mmol) were added and the resulting mixture was stirred at same temperature until the reaction was completed as indicated by TLC. After completion of the reaction, the solvent was removed under vacuum and the residue thus obtained was purified by silica gel flash column chromatography with ethyl acetate/hexane as eluent. Products were confirmed by NMR and LCMS data corresponded to those published.:10). Optical rotation: [α]27D = −71.5 (c 0.5, CH2Cl2).6f 1HNMR (CDCl3, 200 MHz): δ = 2.37–2.45 (m, 5H), 4.37–4.39 (m, 1H), 4.89–5.09 (m, 3H), 5.45–5.53 (m, 1H), 7.09–7.17 (m, 7H), 7.53–7.57 (d, J = 7 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 23.06, 43.47, 58.66, 120.96, 128.15, 128.75, 128.99, 129.97, 130.92, 134.66, 139.03, 141.91, 144.72. TOF-MS (ESI+) calcd [M + Na+] for (C17H19NaNO2S) 324.10, found: 324.40. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 230 nm) tmajor(S) = 19.34 min, tminor(R) = 14.31 min.:12). Optical rotation: [α]27D = −85.4 (c 0.5, CH2Cl2).6f 1H NMR (CDCl3, 200 MHz): δ = 2.39–2.44 (m, 5H), 4.29–4.39 (dd, J = 6.6 and 13.2 Hz, 1H), 5.01–5.09 (m, 3H), 5.38–5.59 (m, 1H), 6.98–7.17 (m, 6H), 7.51–7.55 (d, J = 8 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 21.44, 41.71, 56.56, 119.69, 127.11, 128, 128.42, 129.34, 132.61, 138.82, 143.37. TOF-MS (ESI+) calcd [M + Na+] for (C17H18ClNaNO2S) 358.06, found: 358.23. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 230 nm) tmajor = 22.5 min, tminor = 18.4 min.:12). Optical rotation: [α]27D = −80.8 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ = 2.40–2.43 (t, J = 7 Hz, 2H), 4.37–4.41 (dd, J = 7 and 13.5 Hz, 1H), 4.87–4.88 (d, J = 6 Hz, 1H), 5.06–5.1 (m, 2H), 5.44–5.51 (m, 1H), 6.99–7.02 (m, 2H), 7.12–7.14 (m, 2H), 7.35–7.38 (m, 2H), 7.48–7.51 (m, 1H), 7.64–7.65 (d, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 41.75, 56.42, 119.88, 127.06, 127.97, 128.5, 128.78, 132.49, 132.53, 138.7, 139.49. TOF-MS (ESI+) calcd [M + Na+] for (C16H16ClNaNO2S) 344.05, found: 344.23. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 230 nm) tmajor = 21.75 min, tminor = 18.4 min.:15). Optical rotation: [α]27D = −69.4 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 200 MHz): δ = 2.43–2.49 (t, J = 7 Hz, 2H), 4.44–4.53 (dd, J = 6.4 and 13 Hz, 1H), 5.05–5.17 (m, 3H), 5.44–5.65 (m, 1H), 6.97–7.16 (m, 4H), 7.74–7.78 (d, J = 9 Hz, 2H), 8.16–8.2 (d, J = 9 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 41.66, 56.78, 120.31, 123.9, 128.05, 128.26, 128.67, 132.20, 133.85, 138.01, 146.23, 149.73. TOF-MS (ESI−) calcd [M − H+] for (C16H14ClN2O4S) 365.04, found: 364.82. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 254 nm) tmajor = 17.78 min, tminor = 14.55 min.:16). Optical rotation: [α]27D = −70.7 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 200 MHz): δ = 2.38–2.44 (t, J = 7 Hz, 2H), 4.32–4.42 (dd, J = 6.4 and 13 Hz, 1H), 4.97–5.11 (m, 3H), 5.42–5.59 (m, 1H), 6.93–6.97 (d, J = 8.4 Hz, 2H), 7.3–7.54 (m, 4H), 7.63–7.66 (d, J = 7.4 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 41.68, 56.5, 119.87, 127.04, 128.32, 128.79, 131.44, 132.48, 139.22, 140.2. TOF-MS (ESI+) calcd [M + Na+] for (C16H16BrNaNO2S) 388, found: 388.13. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 230 nm) tmajor = 23.6 min, tminor = 20 min.:15). Optical rotation: [α]27D = −71.5 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ = 2.38 (s, 3H), 2.39–2.43 (m, 2H), 4.34–4.38 (dd, J = 6.5 and 13 Hz, 1H), 5–5.06 (m, 3H), 5.44–5.53 (m, 1H), 6.83–6.87 (m, 2H), 7.03–7.07 (m, 2H), 7.15–7.16 (d, J = 8 Hz, 2H), 7.53–7.54 (d, J = 8.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 21.44, 41.9, 56.4, 115.07, 115.24, 119.6, 127.12, 128.2, 128.26, 129.33, 132.78, 136.12, 137.32, 143.28, 162.95. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 1 mL min−1, λ = 220 nm) tmajor = 17.38 min, tminor = 14.4 min.:12). Optical rotation: [α]27D = −59.4 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 200 MHz): δ = 2.38–2.46 (m, 5H), 4.41–4.51 (dd, J = 6.4 and 12.6 Hz, 1H), 5.04–5.15 (m, 3H), 5.36–5.56 (m, 1H), 7.15–7.32 (m, 4H), 7.53–7.57 (d, J = 8 Hz, 2H), 8.03–8.07 (d, J = 8.6 Hz, 2H). 13C NMR (CDCl3, 25 MHz): δ = 21.45, 41.59, 56.38, 120.49, 123.55, 127.14, 127.56, 129.48, 131.86, 143.82, 147.9. HPLC (Daicel Chiralcel AD-H, hexanes/2-propanol = 95:5, flow rate = 1 mL min−1, λ = 254 nm) tmajor = 22.5 min, tminor = 23.8 min.:18). 1H NMR (CDCl3, 500 MHz): δ = 2.38 (s, 3H), 2.41–2.43 (m, 2H), 4.31–4.4 (dd, J = 6.4 and 13 Hz, 1H), 4.9–4.93 (d, J = 6 Hz, 2H), 5.01–5.09 (m, 2H), 5.39–5.59 (m, 1H), 6.81–6.89 (m, 2H), 7.01–7.17 (m, 4H), 7.51–7.55 (d, J = 8.4 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 22.69, 41.66, 56.56, 20.14, 125.23, 127.05, 127.12, 129.32, 132.31, 143.46. HPLC (Daicel Chiralcel IA, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 220 nm) tmajor = 21.75 min, tminor = 20 min.:12). 1H NMR (CDCl3, 200 MHz): δ = 2.41–2.48 (t, J = 7 Hz, 2H), 2.75 (s, 3H), 4.31–4.4 (dd, J = 6.6 and 13.2 Hz, 1H), 4.8–4.83 (d, J = 5 Hz, 2H), 5.02–5.09 (m, 2H), 5.42–5.62 (m, 1H), 6.67–6.99 (m, 4H), 7.26–7.47 (m, 3H), 7.64–7.68 (d, J = 7.2 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 41.82, 55.21, 56.75, 113.71, 119.09, 127.05, 127.71, 128.66, 132.23, 133.2, 140.49, 158.82. HPLC (Daicel Chiralcel ODH, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 254 nm) tmajor = 28.2 min, tminor = 24.59 min.:16). Optical rotation: [α]27D = −50.1 (c 1, CH2Cl2). 1H NMR (CDCl3, 200 MHz): δ = 2.38–2.44 (m, 5H), 4.31–4.41 (dd, J = 6.6 and 13 Hz, 1H), 4.96–5.12 (m, 3H), 5.4–5.57 (m, 1H), 6.95–7.26 (m, 6H), 7.51–7.55 (d, J = 8.6 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 21.44, 41.71, 56.55, 119.82, 124.84, 126.80, 127.08, 127.47, 129.35, 129.61, 132.53, 134.21, 137.13, 142.23, 143.41. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 220 nm) tmajor = 18.79 min, tminor = 17.14 min.:15). Optical rotation: [α]27D = −46.1 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 200 MHz): δ = 2.36 (s, 3H), 2.42–2.48 (m, 2H), 4.74–4.84 (dd, J = 6.8 and 13.4 Hz, 1H), 5.02–5.14 (m, 3H), 5.38–5.58 (m, 1H), 7.06–7.23 (m, 6H), 7.57–7.21 (d, J = 8 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 21.44, 40.23, 54.07, 119.53, 126.76, 127.15, 128.37, 128.42, 129.31, 132.72, 136.78, 137.78, 143.74. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 230 nm) tmajor = 21.1 min, tminor = 15.1 min.:14). Optical rotation: [α]27D = −48.6 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ = 2.34 (s, 3H), 2.43–2.51 (m, 2H), 4.53–4.64 (dd, J = 7.5 and 14.5 Hz, 1H), 5–5.06 (m, 3H), 5.47–5.55 (m, 1H), 6.82–6.86 (m, 1H), 6.93–6.96 (m, 1H), 7.06–7.15 (m, 4H), 7.54–7.56 (d, J = 8.5 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 21.42, 40.65, 52.92, 115.4, 115.57, 119.26, 123.99, 127.02, 127.33, 127.43, 128.72, 128.95, 129.01, 129.28, 132.87, 137.12, 143.13, 158.97, 160.92. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 1 mL min−1, λ = 220 nm) tmajor = 13.5 min, tminor = 10.63 min.:10). Optical rotation: [α]27D = −80.4 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ = 2.29 (s, 3H), 2.58–2.68 (m, 2H), 4.99–5.01 (d, J = 6.5 Hz, 1H), 5.08–5.13 (m, 2H), 5.2–5.24 (dd, J = 6.5 and 13 Hz, 1H), 5.5–5.58 (m, 1H), 6.99–7 (d, J = 8 Hz, 2H), 7.27–7.3 (m, 2H), 7.45–7.48 (m, 4H), 7.66–7.68 (m, 1H), 7.79–7.81 (m, 1H), 7.89-7.91 (m, 1H). 13C NMR (CDCl3, 125 MHz): δ = 21.37, 41.16, 53.17, 119.44, 122.37, 124.19, 125.04, 125.54, 126.24, 127.06, 127.96, 128.91, 129.12, 133.11, 143.03. TOF-MS (ESI−) calcd [M − H+] for (C21H20NO2S) 350.13, found: 350.19. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 230 nm) tmajor = 23.1 min, tminor = 19.9 min.:12). Optical rotation: [α]27D = −89.1 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ = 2.2 (s, 3H), 2.49–2.6 (m, 2H), 4.53–4.57 (dd, J = 7 and 13.5 Hz, 1H), 5.06–5.1 (m, 3H), 5.47–5.58 (m, 1H), 6.96–6.97 (d, J = 8 Hz, 2H), 7.18–7.2 (m, 1H), 7.42–7.43 (m, 3H), 7.49–7.51 (d, J = 8 Hz, 2H), 7.63–7.65 (m, 2H), 7.74–7.76 (m, 1H). 13C NMR (CDCl3, 125 MHz): δ = 21.25, 41.67, 57.30, 119.37, 124.22, 125.82, 125.89, 126.07, 127.09, 127.47, 127.78, 128.25, 129.13, 132.64, 132.94, 133, 137.31, 143.08. TOF-MS (ESI−) calcd [M − H+] for (C21H20NO2S) 350.13, found: 350.20. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 95:5, flow rate = 0.8 mL min−1, λ = 220 nm) tmajor = 27.9 min, tminor = 20.8 min.:10). Optical rotation: [α]27D = −90.5 (c 0.6, CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ = 2.32–2.43 (m, 5H), 4.03–4.04 (m, 1H), 4.85–4.86 (d, J = 6.5 Hz, 1H), 5.05–5.11 (m, 2H), 5.61–5.69 (m, 1H), 5.78–5.62 (m, 1H), 6.27–6.3 (m, 1H), 7.12–7.25 (m, 7H), 7.72–7.74 (d, J = 8 Hz, 2H). 13C NMR (CDCl3, 125 MHz): δ = 21.36, 40.14, 55.18, 119.33, 126.34, 127.29, 127.66, 127.92, 128.04, 128.10, 128.28, 128.35, 128.6, 128.71, 128.93, 129.47, 129.69, 129.78, 131.53, 132.77, 136.14, 137.91, 143.28. TOF-MS (ESI−) calcd [M − H+] for (C19H20NO2S) 326.13, found: 326.23. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 96:4, flow rate = 0.8 mL min−1, λ = 254 nm) tmajor = 24.5 min, tminor = 26.05 min.:11). Optical rotation: [α]27D = −94.5 (c 0.5, CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ = 1.56 (s, 3H), 2.28–2.35 (m, 2H), 2.37 (s, 3H), 3.91–3.95 (dd, J = 6.5 and 13.5 Hz, 1H), 4.62–4.63 (d, J = 6 Hz, 1H), 5.09–5.12 (m, 2H), 5.59–5.67 (m, 1H), 6.25 (s, 1H), 7.01–7.03 (d, J = 7.5 Hz, 1H), 7.18–7.22 (m, 3H), 7.28–7.29 (m, 2H), 7.71–7.72 (d, J = 8.5 Hz, 1H). 13C NMR (CDCl3, 125 MHz): δ = 21.45, 38.5, 60.77, 118.92, 126.56, 127.41, 127.95, 128.2, 128.83, 129.39, 133.24, 135.38, 136.94, 137.65, 143.22. TOF-MS (ESI−) calcd [M − H+] for (C20H23NO2S) 340.14, found: 340.35. HPLC (Daicel Chiralcel OD-H, hexanes/2-propanol = 96:4, flow rate = 0.8 mL min−1, λ = 254 nm) tmajor = 18.96 min, tminor = 20.33 min.The crude aldehyde was dissolved in tert-butanol (700 μL) and 2-methyl-2-butene (0.96 mmol, 100 μL) was added. A solution of NaClO2 (0.90 mmol, 82 mg) and NaH2PO4 (0.78 mmol, 93 mg) in H2O (300 μL) was then slowly added. After stirring the reaction for 12 h at room temperature, conc. HCl (50 μL) was added drop wise and the reaction mixture was extracted with EtOAc. The organic layer was concentrated to yield the crude product which was then purified by column chromatography (1:5, MeOH:DCM as the eluent) to give the desired compound 7 as a white solid. Optical rotation: [α]27D = −30.5 (c 0.3, CH2Cl2). 1H NMR (CDCl3, 500 MHz): δ = 2.36 (s, 3H), 2.78–2.93 (m, 2H), 4.7–4.74 (m, 1H), 5.89–5.9 (d, J = 8 Hz, 1H), 7.09–7.18 (m, 7H), 7.57–7.58 (m, 2H). 13C NMR (CDCl3, 125 MHz): δ = 21.46, 40.87, 53.99, 126.44, 127.06, 127.84, 128.6, 129.47, 137.05, 138.95, 143.39, 175.22.
Acknowledgements
CSIR-CSMCRI registration no is 122/2014. Authors are thankful to CSIR and CSIR-Indus Magic Project CSC0123 for financial support. Debashis Ghosh is thankful to AcSIR for Ph. D enrolment. Analytical Discipline and Centralized Instrumental Facility is gratefully acknowledged for providing instrumental facilities.Notes and references
- (a) K. Furuta, M. Mouri and H. Yamamoto, Synlett, 1991, 561–562 CrossRef CAS PubMed; (b) G. E. Keck, K. H. Tarbet and L. S. Geraci, J. Am. Chem. Soc., 1993, 115, 8467 CrossRef CAS; (c) C. M. Yu, S. K. Yoon, H. S. Choi and K. Baek, Chem. Commun., 1997, 763 RSC; (d) K. M. Waltz, J. Gavenonis and P. J. Walsh, Angew. Chem., Int. Ed., 2002, 41, 3697–3699 CrossRef CAS; (e) S. Kobayashi, C. Ogawa, H. Konishi and M. Sugiura, J. Am. Chem. Soc., 2003, 125, 6610–6611 CrossRef CAS PubMed; (f) A. V. Malkov, M. Bell, M. Orsini, D. Pernazza, A. Massa, P. Herrmann, P. Meghani and P. Kočovský, J. Org. Chem., 2003, 68, 9659–9668 CrossRef CAS PubMed; (g) A. V. Malkov, L. Dufková, L. Farrugia and P. Kočovský, Angew. Chem., Int. Ed., 2003, 42, 3674–3677 CrossRef CAS PubMed; (h) H. Hanawa, T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 1708–1709 CrossRef CAS PubMed; (i) G. J. Rowlands and W. K. Barnes, Chem. Commun., 2003, 2712–2713 RSC; (j) A. Kina, T. Shimada and T. Hayashi, Adv. Synth. Catal., 2004, 346, 1169–1174 CrossRef CAS; (k) J. G. Kim, K. M. Waltz, I. F. Garcia, D. Kwiatkowski and P. J. Walsh, J. Am. Chem. Soc., 2004, 126, 12580–12585 CrossRef CAS PubMed; (l) Y.-C. Teo, K.-T. Tan and T.-P. Loh, Chem. Commun., 2005, 1318–1320 RSC; (m) R. Takita, K. Yakura, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 13760–13761 CrossRef CAS PubMed; (n) M. Nakajima, S. Kotani, T. Ishizuka and S. Hashimoto, Tetrahedron Lett., 2005, 46, 157–159 CrossRef CAS PubMed; (o) J. F. Traverse, Y. Zhao, A. H. Hoveyda and M. L. Snapper, Org. Lett., 2005, 7, 3151–3154 CrossRef CAS PubMed; (p) S. E. Denmark, J. Fu, D. M. Coe, X. Su, N. E. Pratt and B. D. Griedel, J. Org. Chem., 2006, 71, 1513–1522 CrossRef CAS PubMed; (q) X. Zhang, D. Chen, X. Liu and X. Feng, J. Org. Chem., 2007, 72, 5227–5233 CrossRef CAS PubMed; (r) A. V. Malkov, P. R. López, L. Biedermannová, L. Rulíšek, L. Dufková, M. Kotora, F. Zhu and P. Kočovský, J. Am. Chem. Soc., 2008, 130, 5341–5348 CrossRef CAS PubMed; (s) J. Huang, J. Wang, X. Chen, Y. Wen, X. Liu and X. Feng, Adv. Synth. Catal., 2008, 350, 287–294 CrossRef CAS; (t) I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6340–6341 CrossRef CAS PubMed; (u) A. Kadlčíková, I. Valterová, L. Ducháčková, J. Roithová and M. Kotora, Chem.–Eur. J., 2010, 16, 9442–9445 CrossRef PubMed; (v) Y. Kuang, X. Liu, L. Chang, M. Wang, L. Lin and X. Feng, Org. Lett., 2011, 13, 3814–3817 CrossRef CAS PubMed; (w) Z. Li, B. Plancq and T. Ollevier, Chem.–Eur. J., 2012, 18, 3144–3147 CrossRef CAS PubMed; (x) B. Bai, L. Shen, J. Ren and H. J. Zhu, Adv. Synth. Catal., 2012, 354, 354–358 CrossRef CAS; (y) D. Ghosh, D. Sahu, S. Saravanan, S. H. R. Abdi, B. Ganguly, N. H. Khan, R. I. Kureshy and H. C. Bajaj, Org. Biomol. Chem., 2013, 11, 3451–3460 RSC.
- Chemical reviews: (a) J. A. Marshall, Chem. Rev., 1996, 96, 31–48 CrossRef CAS PubMed; (b) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed; (c) J. W. J. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732–4739 CrossRef CAS PubMed; (d) R. Ramadhar and R. A. Batey, Synthesis, 2011, 9, 1321–1346 Search PubMed; (e) M. Yus, J. C. G. Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774–7854 CrossRef CAS PubMed.
- For catalytic enantioselective allyl additions to aldimines with stoichiometric amounts of In-based reagents, see: (a) R. Kargbo, Y. Takahashi, S. Bhor, G. R. Cook, G. C. L. Jones and I. R. Shepperson, J. Am. Chem. Soc., 2007, 129, 3846–3847 CrossRef CAS PubMed; (b) K. L. Tan and E. N. Jacobsen, Angew. Chem., Int. Ed., 2007, 46, 1315–1317 CrossRef CAS PubMed . For a related In-catalyzed process involving allylboronates and N-acylhydrazones, see:; (c) A. Chakrabarti, H. Konishi, M. Yamaguchi, U. Schneider and S. Kobayashi, Angew. Chem., Int. Ed., 2010, 49, 1838–1841 CrossRef CAS PubMed.
- For Zn-catalyzed enantioselective reactions of allylborons with N-acyl imines, see: (a) M. Fujita, T. Nagano, U. Schneider, T. Hamada, C. Ogawa and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 2914–2915 CrossRef CAS PubMed; (b) S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2007, 129, 15398–15404 CrossRef CAS PubMed.
- For Ag-catalyzed allyl addition to aldimines, see: M. Naodovic, M. Wadamoto and H. Yamamoto, Eur. J. Org. Chem., 2009, 30, 5129–5131 CrossRef PubMed.
- For Pd-catalyzed enantioselective allylation of imines, see: (a) H. Nakamura, K. Nakamura and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 4242–4243 CrossRef CAS; (b) R. A. Fernandes, A. Stimac and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 14133–14139 CrossRef CAS PubMed; (c) R. A. Fernandes and Y. Yamamoto, J. Org. Chem., 2004, 69, 735–738 CrossRef CAS PubMed; (d) O. A. Wallner and K. J. Szabó, Chem.–Eur. J., 2006, 12, 6976–6983 CrossRef CAS PubMed; (e) J. Aydin, K. S. Kumar, M. J. Sayah, O. A. Wallner and K. J. Szabó, J. Org. Chem., 2007, 72, 4691–4697 CrossRef PubMed; (f) X.-C. Qiao, S.-F. Zhu, W.-Q. Chen and Q.-L. Zhou, Tetrahedron: Asymmetry, 2010, 21, 1216–1220 CrossRef CAS PubMed; (g) R. A. Fernandes and J. L. Nallasivam, Org. Biomol. Chem., 2012, 10, 7789–7800 RSC; (h) R. A. Fernandes and D. A. Chaudhari, Eur. J. Org. Chem., 2012, 1945–1952 CrossRef CAS.
- For Cu-catalyzed allyl additions to R-amino esters, see: (a) With allylstannanes: X. Fang, M. Johannsen, S. Yao, N. Gathergood, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 1999, 64, 4844–4849 CrossRef CAS PubMed; (b) With alkenes: D. Ferraris, B. Young, C. Cox, T. Dudding, W. J. Drury III, L. Ryzhkov, A. E. Taggi and T. Lectka, J. Am. Chem. Soc., 2002, 124, 67–77 CrossRef CAS PubMed; (c) With allylsilanes: H. Kiyohara, Y. Nakamura, R. Matsubara and S. Kobayashi, Angew. Chem., Int. Ed., 2006, 45, 1615–1617 CrossRef CAS PubMed; (d) With allylborons: E. M. Vieira, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 3332–3335 CrossRef CAS PubMed.
- H. B. Hepburn and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 11605–11610 CrossRef CAS PubMed.
- (a) D. Enders and U. Reinhold, Tetrahedron: Asymmetry, 1997, 8, 1895–1946 CrossRef CAS; (b) R. Bloch, Chem. Rev., 1998, 98, 1407–1438 CrossRef CAS PubMed; (c) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069–1094 CrossRef CAS PubMed; (d) H. Ding and G. K. Friestad, Synthesis, 2005, 2815–2829 CAS; (e) G. K. Friestad and A. K. Mathies, Tetrahedron, 2007, 63, 2541–2569 CrossRef CAS PubMed.
- R. I. Kureshy, K. J. Prathap, S. Agrawal, N. H. Khan, S. H. R. Abdi and R. V. Jasra, Eur. J. Org. Chem., 2008, 3118–3128 CrossRef CAS.
- (a) P. Kumari, P. K. Bera, N. H. Khan, R. I. Kureshy, S. H. R. Abdi and H. C. Bajaj, Catal. Sci. Technol., 2014, 4, 563–568 RSC; (b) J. C. Byun, D. H. Mun and K.-M. Park, Bull. Korean Chem. Soc., 2014, 35, 269–272 CrossRef CAS; (c) H. S. Jena, New J. Chem., 2014, 38, 2486–2499 RSC.
- The details characterizations of the ligands were described elsewere.
- (a) S. Gandhi and B. List, Angew. Chem., Int. Ed., 2013, 52, 2573–2576 CrossRef CAS PubMed; (b) Enantioselective Synthesis of β-amino acids, ed. E. Juaristi and V. A. Soloshonok, Wiley-VCH, New York, 2nd edn, 2005 Search PubMed; (c) M. Liu and M. P. Sibi, Tetrahedron, 2002, 58, 7991–8035 CrossRef CAS.
- (a) K. R. Guertin and Y.-M. Choi, Curr. Med. Chem., 2007, 14, 2471–2481 CrossRef CAS; (b) K. B. Hansen, J. Balsells, S. Dreher, Y. Hsiao, M. Kubryk, M. Paluki, N. Rivera, D. Steinhuebel, J. D. Armstron-g III, D. Askin and E. J. J. Grabowski, Org. Process Res. Dev., 2005, 9, 634–639 CrossRef CAS; (c) N. Yasuda, Y. Hsiao, M. S. Jensen, N. R. Rivera, C. Yang, K. M. Wells, J. Yau, M. Paluki, L. Tan, P. G. Dormer, R. P. Volante, D. L. Hughes and P. J. Reider, J. Org. Chem., 2004, 69, 1959–1966 CrossRef CAS PubMed; (d) E. K. Rowinsky, Annu. Rev. Med., 1997, 48, 353–374 CrossRef CAS PubMed; (e) F. Hessler, A. Korotvička, D. Nečas, I. Valterová and M. Kotora, Eur. J. Org. Chem., 2014, 2543–2548 CrossRef CAS.
- For reviews on β-peptides, see: (a) W. S. Horne and S. M. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS PubMed; (b) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS PubMed; (c) D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS PubMed; (d) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed; (e) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon, New York, 1981 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10929e |
| This journal is © The Royal Society of Chemistry 2014 |