
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA universal route to improving conjugated macromolecule photostability†
H. Santos
Silva
ab,
A.
Tournebize
cde,
D.
Bégué
*a,
H.
Peisert
c,
T.
Chassé
c,
J.-L.
Gardette
de,
S.
Therias
de,
A.
Rivaton
de and
R. C.
Hiorns
*f
aUniversité de Pau et des Pays de l'Adour (UPPA), IPREM ECP, CNRS-UMR 5254, 2 Avenue Président Angot, 64053 Pau, France. E-mail: didier.begue@univ-pau.fr
bUPPA, IPREM EPCP, CNRS-UMR 5254, 64053 Pau, France
cInstitute for Physical and Theoretical Chemistry, Eberhard-Karls-University, Auf der Morgenstelle 18, D-72076 Tübingen, Germany
dUniversité Blaise Pascal, Institut de Chimie de Clermont-Ferrand, Equipe Photochimie, BP 10448, F-63000 Clermont-Ferrand, France
eCNRS, UMR 6296, ICCF, Équipe Photochimie, BP 80026, F-63171 Aubière, France
fCNRS, IPREM EPCP, CNRS-UMR 5254, 64053 Pau, France. E-mail: roger.hiorns@univ-pau.fr
First published on 16th October 2014
Abstract
This predictive study demonstrates that the introduction of aromatic-oxy-alkyl links surprisingly makes materials more resistant to photo-oxidative degradation by reducing hydrogen abstraction. This revelation makes it possible, for the first time, to design a toolbox of substituents for soluble, photostable conjugated materials.
Wide-ranging strategies to modify conjugated organic molecules and polymers for target applications in organic electronics,1 organic photovoltaics (OPVs),2 non-linear optics,3 bio-interfacing,4 and chemical sensors,5 are performed with the aim of understanding structure–efficiency relationships,6 but also increasing material stabilities,7,8 processibilities and cost effectiveness.9,10 However, organic materials suffer catastrophic degradation under light in combination with atmospheric oxygen.11 If an efficient material is unstable, its use is constrained. To limit the process of photo-oxidation, additives such as UV-screeners and antioxidants are thus often essential in commercial polymer applications.12 Unfortunately, these strategies are not available for conjugated polymers in organic electronics and opto-electronics. For example, in OPVs, UV-screeners reduce efficiencies and anti-oxidants can act as charge traps. Therefore, it is necessary to design intrinsically stable materials.
The mechanisms by which most macromolecules degrade in air on exposure to UV-visible light are well understood.13–15 These mechanisms apply to both classic commodity polymers and modern low bandgap polymers. The radical chain oxidation of a polymer (PH) is due to hydrogen abstraction by a free-radical (r˙), itself formed from the photonic excitation of a chromophore as in:
| PH + r˙ → P˙ + rH |
In the propagation step, the macroradical (P˙) reacts with atmospheric oxygen to give a peroxy radical (PO2˙) which in turn abstracts another labile hydrogen to yield a hydroperoxide. The latter thermally or photochemically decomposes to furnish macroalkoxy PO˙ and hydroxyl radicals.8,12,14,15 Depending on its structure, a macroalkoxy radical PO˙ decomposes, which variously impact upon the polymer properties.12 The lability of polymer hydrogens towards abstraction is thus a key point in terms of the durability of material properties.
In order to make conjugated polymers soluble in organic solvents for facile processing, side-chains are required. However, studies have unambiguously shown that the aforementioned hydrogen abstraction most likely occurs at the side-chains, notably sites alpha to the conjugated backbone.8,16 It is therefore crucial to investigate the effect of the structure of the side-chain on the stability of the final material.
The vast majority of conjugated polymers have carbochains, either branched or linear,17 as side-groups, as shown in Fig. 1a. While oxygen has been used in side-chains, it has not been for reasons of stability but rather for solubility, synthetic facility and its ability to act as a charge donor, increasing the density of states of the macromolecule.18 Generally, chemists have tended to shun using an ether link, as in Fig. 1b, because it was believed, like in Fig. 1c, that it would increase degradation rates. This was because prior work indicated that conjugated aromatic-oxy-alkyl links enhanced photo-oxidation (Scheme S1, ESI†) in simple analogy to the known behaviour of all-aliphatic systems (e.g., polyethylene versus poly(ethylene oxide) (PEO), Fig. S1, ESI†).19
 | ||
| Fig. 1 Chemical groups used in the discussion: (a) aromatic-alkyl; (b) aromatic-oxy-alkyl; and (c) aromatic-alkyl-oxy-alkyl. | ||
This work finds the exceptional revelation that the insertion of an oxygen into conjugated and non-conjugated aromatic-alkyl polymers at the position shown in Fig. 1b can stabilize polymers against photo-oxidation. Furthermore, we establish which parameters are of importance in prescribing stabile structures towards photo-oxidation, namely: bond dissociation energies (EBD);20 stabilities of product macro-radicals;20,21 and hydrogen labilities in terms of the delocalization of remaining macrostructural radicals. Additional but discarded parameters are those of bond dipole moments (BDMs) (Table S1, ESI†), and transition state geometries (Fig. S2, ESI†).22 In the former case the results were found irrelevant due to the radical (non-polar) nature of the degradation. In the latter, it was impossible to be assured of pinpointing the lowest transition state in 3-dimensions.
Molecular geometries of model compounds were fully optimized within Density Functional Theory (DFT) and Hartree–Fock (HF) methods, using a double-ζ polarized basis-set (6-31G**).23,24 DFT calculations used a B3LYP exchange–correlation functional.25 Where required, the open-shell wave function was set to an unrestricted type (UHF/UKS). Modeling of hydrogen-abstracted geometries was performed with a doublet multiplicity in a neutral state. BDMs were calculated with respect to the ground state. All calculations were performed using Orca 2.9 software.26 Spin distribution was extracted from the Löwdin partition.27 Corroborating DFT results (Table S2, ESI†), HF results can be found in Fig. S3 and S4, and Table S3, ESI.†
The model compounds depicted in Fig. 2 are used as they permit calculations over a variety of typical hydrogen positions. The conclusions drawn from these models were then tested on the range of structures shown in Fig. 3, which include oligophenylenes, oligothiophenes, oligo-TBT and oligo-BT7. The results of these studies showed that the conclusions could be extended to medium and low-band gap conjugated polymers and, furthermore to future designs.
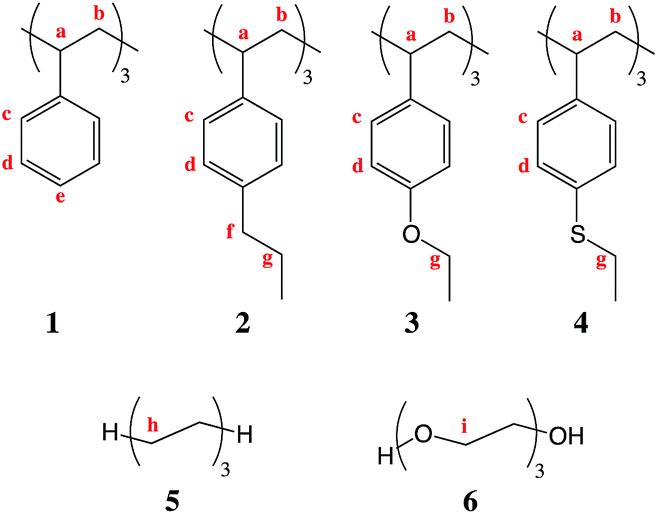 | ||
| Fig. 2 Basic molecular structures used in this study. | ||
 | ||
| Fig. 3 Additional chemical structures used in this study where n = 1 to 4. | ||
Molecules 1 to 4 are oligostyrenes, and 5 and 6 are oligo(ethylene) and oligo(oxyethylene), respectively. While 1 provided a standard, 4 was calculated to deduce effects from electronegativity while retaining the chalcogenic character of the heteroatom. EBD values of the α-hydrogens to the aromatic group, or nearest neighbor equivalents to the oxygen atom, were calculated along with the thermodynamic stabilities of the final macroradical compound formed after hydrogen abstraction. This was performed on the basis that the mechanism of degradation is initiated by hydrogen abstraction by a photo-generated radical, such that:
| EBD = (EMRT + EHRT) − EMST |
| Hydrogen label | Bond dissociation energy (kcal mol−1) | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| a | 85.35 | 86.17 | 86.98 | 88.98 | — | — |
| b | — | 101.36 | 101.29 | 101.90 | ||
| f | — | 92.79 | — | — | — | — |
| g | — | 104.74 | 100.45 | 98.84 | — | — |
| h | — | — | — | — | 104.80 | — |
| i | — | — | — | — | — | 101.56 |
Comparing 5 and 6, one notes that the presence of an oxygen atom makes the attached hydrogen more labile; the energetic difference is around 3 kcal mol−1. As expected oxygen favours H-abstraction from adjacent methylene groups in all-aliphatic systems.28 Then looking at molecules 1–4, and excluding the tertiary C–Hs (labelled ‘a’) as they are expectedly weak, it is extraordinary that oxygen has a protecting effect on the nearest hydrogen. The weakest C–H bond is now no longer 92.79 kcal mol−1 (2f) but 100.45 kcal mol−1 (3g); a gain in energy of around 8 kcal mol−1 has been made. Sulfur has a similar impact, raising the lowest energy C–H bond by around 6 kcal mol−1. Furthermore the problem has not simply been moved along the alkylated chain. Finally as mentioned above, the EBD values of 1–4a, as expected, are all very low due to the tertiary carbon. Interestingly though, oxygen and sulfur slightly stabilise this group. No discernable affect is accorded to hydrogens 2b–4b.
In order to better understand the underlying effects, we turned again to the primary model set to consider the stabilization of the formed macroradical in our primary models. Such stabilities should test and, if correct, confirm the unexpected heteroatomic stabilization. Calculations were made for overall energy gains for RH + OH˙ → R˙ + H2O; hydrogens were abstracted from the weakest points on the aromatic-alkyl and aromatic-oxy-alkyl chains (RH). Once performed, confirmation of the results was found. Compound 2 (2f abstraction) gives rise to the most stable structure i.e., the formation of a radical is favoured, whereas the aromatic-oxy-alkyl radical of 3 (3g abstraction) does not favour radical formation. For the wholly aliphatic molecules 5 and 6, results again confirm the destabilizing effect of oxygen, i.e., the macroradical of 5 is less stable and thus less favoured than that of 6 (respectively, −16.74 and −20.63 kcal mol−1) (Table 2).
| Thermodynamic stability of R˙ (kcal mol−1) | |||
|---|---|---|---|
| 2f | 3g | 5h | 6i |
| −28.97 | −19.82 | −16.74 | −20.63 |
To better understand the stability of aromatic-oxy-alkyl moieties and instability of alkyl-oxy-alkyl groups, we determined the spatial delocalization of unshared electrons in the macroradicals of 1–6 (see also Table S4, ESI†). In each case, the lowest energy hydrogen (outside of tertiary hydrogens) was removed. In 5 the unpaired electron is highly localized at the carbon from which the hydrogen was abstracted, with a partition coefficient of 0.88. However, in the oxygenated molecule 6, this value is reduced to 0.77; the adjacent oxygen assumes a coefficient of 0.15. This corroborates the above results as the hydrogen is more labile (more unstable) because its radical product is delocalized away from the native carbon. However, for compounds 2 and 3, we find that in the former, the coefficients for the unpaired electron are 0.63 at the native carbon and 0.35 over the aromatic ring, and in the latter 0.77 on the native carbon, 0.12 on oxygen, and only 0.04 at the ring. In other words, hydrogen abstraction at 2f results in a more stabilized macroradical than that from 3g. In effect, the oxygen in 3 has blocked the unpaired electron from moving towards the aromatic group, with the result that macroradical is less stabilized, and hydrogen abstraction is less favoured. In the aliphatic structures, 5 displays a more localized electronic behaviour because the oxygen in 6 takes up some of the unpaired electron presence. However, in aromatic structures, this effect gives rise to the opposite result: the oxygen takes part in delocalizing the electron but blocks its further delocalization to the aromatic ring; this makes the radical product less stable and less likely to be formed.
To demonstrate that the insertion of oxygen into aromatic-alkyl bonds has a universal effect, we turned to the 32 chemical structures in Fig. 3. All EBD values were calculated, and the weakest bonds have their energies listed in Table 3. In all cases, the introduction of oxygen increases C–H bond strengths. The values, across a range of structures, indicate that the process is based on the local character of the molecule, regardless of: the presence of heteroatoms in the conjugated backbone; or conjugation length. Furthermore, oligomers without oxy-moieties generally show a weakening of bond strength with increasing aromatic conjugation, an effect not observed for oxygenated structures. This further corroborates that the ether link blocks unshared electron delocalization. By extension, the insertion of oxygen is a powerful tool for stabilization of conjugated polymers.
| E BD (kcal mol−1) | ||||
|---|---|---|---|---|
| n = 1 | n = 2 | n = 3 | n = 4 | |
| Oligo(1,4-dialkyl)phenylene | −92.83 | −92.87 | −94.81 | −95.01 |
| Oligo(1,4-dialkoxy)phenylene | −100.55 | −100.34 | −100.21 | −100.21 |
| Oligo[(3-alkyl)thiophene] | −92.81 | −90.95 | −90.22 | −86.94 |
| Oligo[(3-alkoxy)thiophene] | −100.67 | −100.61 | −100.90 | −100.97 |
| Oligo(alkylTBT) | −90.39 | −93.19 | −92.55 | −92.51 |
| Oligo(alkoxyTBT) | −97.42 | −100.26 | −100.50 | −102.54 |
| Oligo(alkylTB7) | −87.53 | −92.32 | −91.99 | −92.08 |
| Oligo(alkoxyTB7) | −100.45 | −100.59 | −100.62 | −100.73 |
Conclusions
An oxygen atom attached to an sp3 carbon decreases the stability of a macromolecule towards oxidative photodegradation, as demonstrated by the known behaviors of PE and PEO. However, and in contrast to expectations, an aromatic-oxy-alkyl moiety is more resistance to photo-oxidation than an aromatic-alkyl group. The effect of oxygen is a localized one. It is its position that determines how stability is impacted: an oxygen atom adjacent to an aromatic group can block electronic communication through its relatively high electronegativity; adjacent to an aliphatic chain, it diminishes stability by spreading unpaired electrons. Alkyl side-chains that are not protected from aromatic groups by oxygen see their bond strength reduced as aromatic conjugation is increased. This is an important result for chemists preparing conjugated polymers for a wide variety of applications where long-term photo-oxidative stability is required. It is important to note also that this effect is neither strongly dependent on the conjugate polymer structure nor on the presence of heteroatoms. While it should be noted that other effects need to be taken into account when designing new materials for applications, such as the generation of singlet oxygen by the polymer when in combination with other materials,29 it is expected that these new design rules will enhance long term polymer stability.Acknowledgements
The research leading to these results has received funding from European Union Seventh Framework Program (FP7/2011) under grant agreement ESTABLIS no. 290022. Dr M. Pédeutour is thanked for administrative support.Notes and references
- (a) X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef CAS PubMed; (b) A. Pron, P. Gawrys, M. Zagorska, D. Djurado and R. Demadrille, Chem. Soc. Rev., 2010, 39, 2577 RSC.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed.
- D. L. Elder, S. J. Benight, J. Song, B. H. Robinson and L. R. Dalton, Chem. Mater., 2014, 26, 872 CrossRef CAS.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679 CrossRef CAS.
- A. Malhotra, M. McInnis, J. Anderson and L. Zhai, in Intelligent Stimuli-Responsive Materials, ed. Q. Li, John Wiley & Sons Inc., New Jersey, 2013, ch. 13 Search PubMed.
- P. M. Beaujuge, H. N. Tsao, M. R. Hansen, C. M. Amb, R. Chad, J. Subbiah, K. R. Choudhury, A. Mavrinsky, W. Pisula, J.-L. Brédas, F. So, K. Müllen and J. R. Reynold, J. Am. Chem. Soc., 2012, 134, 8944 CrossRef CAS PubMed.
- (a) X. Wu, X. Sun, Z. Guo, J. Tang, Y. Shen, T. D. James, H. Tian and W. J. Zhu, J. Am. Chem. Soc., 2014, 136, 3579 CrossRef CAS PubMed; (b) S. Bellani, D. Fazzi, P. Bruno, E. Giussani, E. V. Canesi, G. Lanzani and M. R. Antognazza, J. Phys. Chem. C, 2014, 118, 6291 CrossRef CAS.
- (a) A. Tournebize, P.-O. Bussière, P. Wong-Wah-Chung, A. Rivaton, J.-L. Gardette, S. Beaupré and M. Leclerc, Adv. Energy Mater., 2013, 3, 478 CrossRef CAS; (b) A. Tournebize, A. Rivaton, J.-L. Gardette, C. Lombard, B. Pépin-Donat, S. Beaupré and M. Leclerc, Adv. Energy Mater., 2014 DOI:10.1002/aenm.201301530; (c) A. Rivaton, A. Tournebize, J. Gaume, P.-O. Bussière, J.-L. Gardette and S. Therias, Polym. Int., 2013, 63, 1335 CrossRef.
- M. Manceau, E. Bundgaard, J. E. Carlé, O. Hagemann, M. Helgesen, R. Søndergaard, M. Jøgensen and F. C. Krebs, J. Mater. Chem., 2011, 21, 4132 RSC.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686 CrossRef PubMed.
- (a) S. H. Hamid, M. B. Amin and A. G. Maadhah, Handbook of Polymer Degradation, Marcel Dekker, Inc., New York, 1992 Search PubMed; (b) J. F. Rabek, Photodegradation of Polymers, Springer, Berlin, 1996 Search PubMed.
- J.-L. Gardette, Fundamental and technical aspects of the photooxidation of polymers, in Handbook of Polymer Science, ed. S. H. Hamid, Marcel Dekker Inc., 2000, p. 671 Search PubMed.
- M. Gardette, A. Perthue, J.-L. Gardette, T. Janecska, E. Foldes, B. Pukanszky and S. Therias, Polym. Degrad. Stab., 2013, 98, 2383 CrossRef CAS PubMed.
- (a) M. Manceau, A. Rivaton, J.-L. Gardette, S. Guillerez and N. Lemaître, Polym. Degrad. Stab., 2009, 94, 898 CrossRef CAS PubMed; (b) A. Tournebize, P.-O. Bussière, A. Rivaton, J.-L. Gardette, H. Medlej, R. C. Hiorns, C. Dagron-Lartigau, F. C. Krebs and K. Norrman, Chem. Mater., 2013, 25, 4522 CrossRef CAS.
- (a) J. L. Bolland and G. Gee, Trans. Faraday Soc., 1946, 42, 236 RSC; (b) L. Audouin, V. Langlois, J. Verdu and J. C. M. de Bruin, J. Mater. Sci., 1994, 29, 569 CrossRef CAS.
- A. Rivaton, M. Manceau, S. Chambon, J.-L. Gardette, S. Guillerez and N. Lemaître, Polym. Degrad. Stab., 2010, 95, 278 CrossRef CAS PubMed.
- (a) N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS PubMed; (b) P.-L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2010, 23, 456 CrossRef.
- (a) B. Van Veller, D. J. Schipper and T. M. J. Swager, J. Am. Chem. Soc., 2012, 134, 7282 CrossRef CAS PubMed; (b) A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed; (c) J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761 CrossRef CAS; (d) G. Koeckelberghs, M. Vangheluwe, C. Samyn, A. Persoons and T. Verbiest, Macromolecules, 2005, 38, 5554 CrossRef CAS; (e) Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135 CrossRef CAS PubMed; (f) R. C. Hiorns, E. Cloutet, E. Ibarboure, A. Khoukh, H. Bejbouji, L. Vignau and H. Cramail, Macromolecules, 2010, 43, 6033 CrossRef CAS; (g) C. Ouhib, A. Dkhissi, P. Iratçabal, R. C. Hiorns, A. Khoukh, J. Desbrières, C. Pouchan and C. Dagron-Lartigau, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7505 CrossRef.
- S. Chambon, A. Rivaton, J.-L. Gardette, M. Firon and L. Lutsen, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 317 CrossRef CAS.
- (a) S. J. Blanksby and G. B. Elisson, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS PubMed; (b) F. Posada, P. Malfreyt and J.-L. Gardette, Comput. Theor. Polym. Sci., 2001, 11, 105 CrossRef CAS; (c) F. Posada, J.-L. Philippart, P. Kappler and J.-L. Gardette, Polym. Degrad. Stab., 1995, 50, 141 CrossRef CAS.
- (a) G. Gryn'ova, K. U. Ingold and M. L. Coote, J. Am. Chem. Soc., 2012, 134, 12979 CrossRef PubMed; (b) I. Rossi, A. Venturini and A. Zedda, J. Am. Chem. Soc., 1999, 121, 7914 CrossRef CAS.
- F. Posada, P. Malfreyt and J.-L. Gardette, Comput. Theor. Polym. Sci., 2001, 11, 95 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed; (b) J. D. Dill and J. A. Pople, J. Chem. Phys., 1975, 62, 2921 CrossRef CAS PubMed; (c) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS PubMed.
- D. Bégué, G. Qiao and C. J. Wentrup, J. Am. Chem. Soc., 2012, 134, 5339 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- F. Neese, The ORCA program system – WIREs, Comput. Mol. Biosci., 2012, 2, 73 CrossRef CAS.
- (a) P.-O. Löwdin, J. Chem. Phys., 1950, 18, 365 CrossRef PubMed; (b) P.-O. Löwdin, Adv. Quantum Chem., 1970, 5, 185 CrossRef.
- S. Morlat and J.-L. Gardette, Polymer, 2001, 42, 6071 CrossRef CAS.
- Y. W. Soon, H. Cho, J. Low, H. Bronstein, I. McCulloch and J. R. Durrant, Chem. Commun., 2013, 49, 1291 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental studies of PEO and PE degradations; EBDs with comparisons of methodologies used. Reconsideration of photo-oxidations of poly(phenylene vinylene) and poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylenevinylene]; BDM calculations. See DOI: 10.1039/c4ra10806j |
| This journal is © The Royal Society of Chemistry 2014 |