
Anion directed conformational diversities of an arene based hexa-amide receptor and recognition of the [F4(H2O)6]4− cluster†
Sourav Chakrabortya,
Ranjan Duttaa,
Bryan M. Wong b and
Pradyut Ghosh*a
b and
Pradyut Ghosh*a
aDepartment of Inorganic Chemistry Indian Association for the Cultivation of Science, 2A & 2B Raja S. C. Mullick Road, Kolkata 700032, India. E-mail: icpg@iacs.res.in; Fax: +91-33-2473-2805
bDepartment of Chemical & Environmental Engineering and Materials Science & Engineering Program, University of California, Riverside, Riverside, California 92521, USA
First published on 13th November 2014
Abstract
A benzene platform based new hexa-amide receptor (L) has shown conformational diversities via isolation of aaabbb (A), aabaab (B) and aaaaaa (C) conformers upon recognition of acetate, nitrate and hydrated-fluoride [F4(H2O)6]4− respectively in the solid state. Solution and DFT calculation studies show fluoride binding selectivity of L.
Receptors based on (tris-2-aminoethylamine) TREN,1 arene,2 cyanuric acid3 and calix pyrrole4 platforms are popular for the recognition of anions. But arene platform based hexapodal receptors are scarcely reported in the anion recognition literature.5 In 1974, Vögtle et al. first reported an octopus like hexapodal ligand that forms a cation complex.6 Later, after thirty four years a DFT calculation study by Allen et al. showed that six alkyl substituted hexa-amides orient in the same direction forming a bowl shaped conformer that binds chloride and triflate with low binding affinity.7 In 2010 our group reported the first example of compartmental recognition of nitrate/acetate by a pentafluorophenyl substituted hexa-amide receptor.8a Later on we have shown recognition of [(F)4(H2O)10]4− cluster in the dimeric capsular assembly of a m-nitrophenyl substituted hexa-amide where all the six arms are in unidirectional conformation.8b Recognition of hydrated fluoride could be a practical approach towards removal of fluoride from water due to its high hydration energy. In principle, hydrated fluoride may exist in various cluster compositions and thus practically could act as guest in fluoride recognition.9a Various fluoride–water clusters have been trapped which includes [F(H2O)]44−,9b [F(H2O)4]−,9c [(F)2(H2O)6]2−,2a [(F)4(H2O)10]4−,8b and [(F)2(H2O)4]2−.2f On the other hand, polymorphism of hexa-host receptors and their solvated inclusion complexes have also been demonstrated.10 Herein we report the single crystal X-ray structural evidence on anion-dependent trapping of unusual conformers of 2-(trifluoromethyl)phenyl substituted hexa-amide by hydrated fluoride (aaaaaaa), nitrate (aabaab) and acetate (aaabbb). Furthermore, we show recognition of a new fluoride–water cluster [(F)4(H2O)6]4− in the dimeric capsular assembly of the hexa-amide along with fluoride binding selectivity.
The hexa-amide receptor L has been synthesized following the previously published procedure (Scheme 1).8 L is crystallized from dioxane–water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) binary solvent mixture in aaabbb conformation (A). Complexation of L with acetate, nitrate and fluoride have resulted complexes 1; [(L)·(CH3COO)2·(TBA)2], 2; [(L)·(NO3)·(TBA)] and 3; [(L)2·(F)4·(H2O)6·(TBA)4] respectively.
:1, v/v) binary solvent mixture in aaabbb conformation (A). Complexation of L with acetate, nitrate and fluoride have resulted complexes 1; [(L)·(CH3COO)2·(TBA)2], 2; [(L)·(NO3)·(TBA)] and 3; [(L)2·(F)4·(H2O)6·(TBA)4] respectively.
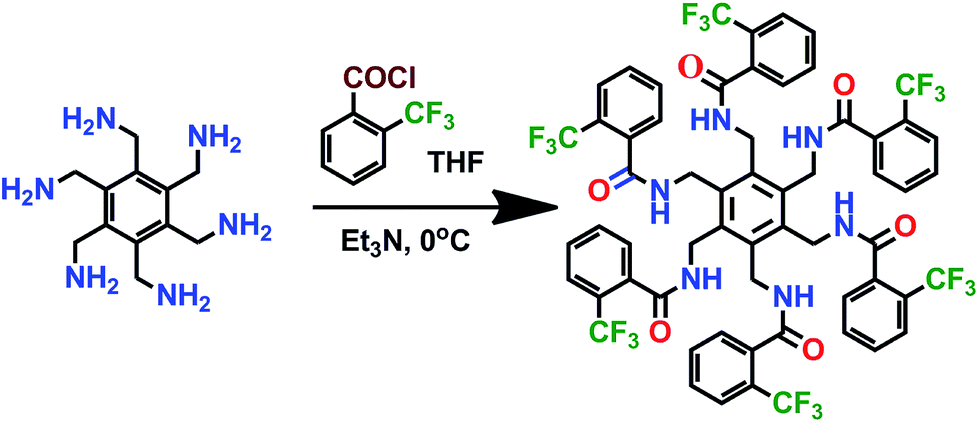 | ||
| Scheme 1 Synthesis of L. | ||
Structural analysis of L has shown orientation of three consecutive arms in one direction versus the other three arms directed towards opposite direction, resulting in a chair-like conformation (aaabbb, conformer A in Fig. 1S, ESI†). The solid state structure of L shows one of the amide protons (N3–H3A) is intra-molecularly hydrogen bonded to the carbonyl oxygen (O1) of another arm and one amide –NH proton is directed towards the cavity composed of three arms (Fig. 16S, ESI†). However, this clearly suggests that two tripodal clefts of hexapodal receptor with aaabbb conformation are complimentary towards compartmental guest recognition. During complexation with acetate (complex 1), all three amide –NH protons are oriented towards the cavity of the tripodal cleft to bind acetate where stoichiometry of binding is found to be 1:2 (host–guest) (Fig. 16S, ESI†). The oxygen atom O2 of acetate is in strong hydrogen bonding interactions with two amide –NH centres N1–H1 and N3–H3 whereas O1 is hydrogen bonded to the third amide centre N2–H2 (Table 3S, ESI†). In complex 1, the distances between the amide nitrogen centres are quite similar to L. Several intermolecular C–H⋯O interactions between acetate and aryl C–H protons of L result in the formation of a 1D-polymeric capsular assembly in complex 1 where the distance between two acetates is 6.8 Å. Similar aaabbb (A) conformer was also observed in case of acetate complexes of pentaflurophenyl8a and m-nitrophenyl8b substituted hexa-amide receptors.
Interestingly, when the guest is changed from acetate to nitrate, L shows different structural conformer in single crystal X-ray study. The nitrate complex of L, i.e. complex 2 [(L)·(NO3)·(TBA)], shows 1:1 (host–guest) recognition where NO3− is recognized in a tetrapodal cleft with overall aabaab (B) conformation (Fig. 1a). Hence, upon changing the guest we have observed that both the host conformation as well as the stoichiometry of binding is altered. In the tetrapodal cleft of complex 2, the NO3− is recognized by four strong N–H⋯O hydrogen bonding interactions from all four amide centres (Table 4S, ESI†). The remaining two arms of L in B conformer point toward opposite direction forming a dipodal cleft without any guest binding. This is an unusual conformation (aabaab) that is definitely stabilized upon recognition of guest like nitrate although same receptor preferred aaabbb conformation in both free ligand and in its acetate complex (1).
 | ||
| Fig. 1 (a) Complex 2 shows recognition of nitrate in unusual B conformation by L; (b) [F4(H2O)6]4− cluster inside the dimeric capsular assembly of L in complex 3 in C conformer. Hydrogen atoms on the phenyl rings are removed for clarity.‡ | ||
Interesting result on the anion directed conformational study is found in case of hydrated fluoride recognition by L in complex 3. Upon changing the guest from planar anions (acetate, nitrate) to spherical anion fluoride, we have observed trapping of very unusual conformer of L i.e. aaaaaa (C) that has recognised [F4(H2O)6]4− in the large cavity of the dimeric capsular assembly of L (Fig. 1b). The asymmetric unit of complex 3 possesses one aaaaaa conformer of L, two fluoride, five water, three dioxane and two TBA counter-cations. The capsular dimension of complex 3 measured from the bridgehead arenes centroid is found to be 11.901 Å. L in complex 3 is able to encapsulate two F− and two water molecules, [F2(H2O)2]2− in its bowl shaped cavity (half capsule). This [F2(H2O)2]2− unit is further hydrogen bonded to another [F2(H2O)2]2− unit through two bridging water molecules (O15) to form [F4(H2O)6]2− cluster in the cavity of a dimeric capsular assembly of L (Fig. 1b). Encapsulation of [F4(H2O)6]4− cluster is assisted by multiple N–H⋯F, O–H⋯F and N–H⋯O strong interactions where the hydrogen bonding distances range from 2.62 Å to 2.81 Å (Table 5S, ESI†). The fluoride ion labelled F16 is hydrogen bonded to amide centres N1–H1 and N2–H2 whereas fluoride ion labelled F17 is hydrogen bonded to amide centres N4–H4 and N5–H5. The other two amide centres namely N3–H3 and N6–H6 are hydrogen bonded to O11 and O14 of [F2(H2O)2]2− unit respectively. Interestingly, the coordination environments of two fluorides (F16 and F17) are different in complex 3. Thus [F4(H2O)6]4− cluster acts as a template to form a dimeric capsular assembly via various hydrogen bonding interactions which orients all the six amide arms in the same direction. However, the unidirectional pattern of all six arms of hexapodal receptors in its native state is very unusual and highly disfavoured in terms of thermodynamics. Thus hydrated fluoride cluster traps one of the most unfavourable conformers in case of arene platform based hexa-amide receptor. It is noteworthy to mention that recognition of [(F)4(H2O)10]4− cluster in the dimeric capsular assembly of a m-nitrophenyl substituted hexa-amide in aaaaaa (C) conformation was previously reported by our group.8b
DFT studies have been employed to find the binding energies of these three anion complexes (1, 2 & 3). Table 8S (ESI†) summarizes the binding energies for the complexes calculated at the M06-2X/6-31G(d,p) level of theory. Among all three complexes, complex 3 (136.50 kcal mol−1) have the largest binding energy where each of the six arms of hexa-amides are in the aaaaaa (C) conformation to form strong hydrogen bond with a fluoride ion in the center of the complex. It is important to note that all of these calculations were carried out in the gas phase, and the large binding energy arises from the strong columbic attraction of the negatively-charged anion with the hydrogen atoms of the receptor. The presence of explicit solvent molecules would effectively screen the charge of the anion and lower the binding energy; however, the size of these complexes is too prohibitively large to include explicit solvent. Next in the line of decreasing binding energies is complex 1 (77.44 kcal mol−1) with aaabbb conformation (A) where three arms of the ligands are hydrogen bonded to an acetate anion. On a mechanistic level, the binding energy of complex 1 is smaller than the binding energy for complexes 3 for two reasons: (i) complex 1 only involves the interaction of three ligand functional groups with the anion in the aaabbb conformation, whereas complex 3 involves attractive interactions with all six arms in the aaaaaa conformation; (ii) the fluoride anion is significantly more electronegative than the acetate ion, leading to a much higher binding energy for complex 3. Interestingly, DFT calculation studies on our previously reported [(F)4(H2O)10]4− cluster encapsulation by m-nitrophenyl substituted hexa-amide receptor8b showed binding energy of 136.86 kcal mol−1 (Fig. 21S and Table 12S, ESI†) which is almost equal in magnitude with complex 3. Following the previously discussed systems, complex 2 possess the next binding energy (71.09 kcal mol−1) and involves in hydrogen bonding interactions with a nitrate anion in aabaab (B) conformation. The details of the coordinates and optimized structures are mentioned in ESI (Fig. 18S–21S, Tables 9S–11S†).
Anion binding propensities of the receptor is evaluated by Isothermal Titration Calorimetry (ITC) and 1H-NMR titration studies. Qualitative 1H-NMR studies of anions (F−, Cl−, Br−, I−, NO3−, CH3COO−) with L are carried out in DMSO-d6 which reveals only binding of F− and CH3COO− in solution (Fig. 14S, ESI†). Both acetate and fluoride have shown 1:2 binding stoichiometry which is evident from the Job's plot analysis which supports the solid state binding pattern. L has shown binding with acetate with downfield shift of –NH proton upto 0.565 ppm (Fig. 13S, ESI†). Acetate shows the association constant 5.97 × 105 M−2 (Table 7S, ESI†). During the binding constant estimation of fluoride, we have monitored the shift of aromatic –CH protons as broadening and subsequent disappearance of amide –NH signal is observed. The association constant estimated by 1H-NMR titration for fluoride is found to 4.14 × 107 M−2 with upfield shift of –CH proton up to 0.091 ppm (Fig. 2a and b). Measureable data in ITC study are only obtained for acetate with L in DMSO (Fig. 2c). Unfortunately, binding of TBANO3 with hexa-amides in DMSO turned out to be too weak to be reliably quantified by ITC measurements. In the case of fluoride the ITC profiles have shown fitting in a 1:2 (sequential sites) model with high Chi2/DOF values and thus are not included. The thermodynamic and kinetic parameters associated with acetate to L binding obtained from both ITC and 1H-NMR titration studies are tabulated in Tables 6S and 7S, ESI.† L has shown exothermic binding pattern towards acetate binding (Fig. 2c). This titration data fit well to a sequential binding model where binding sites are taken as two. During titration with acetate we have observed no heat pulse beyond 2 equivalents of anions that also confirms the choice of model for data fitting. The binding of the first acetate is found to be entropy (ΔH1 = −1795 cal mol−1, ΔS1 = 10.5 cal mol−1 deg−1) driven whereas binding of the second acetate is enthalpy (ΔH2 = −2360 cal mol−1, ΔS2 = 2.92 cal mol−1 deg−1) driven. The association constant estimated for acetate binding is 9.76 × 105 M−2 which invariably matched with the 1H-NMR data. The solution state studies have shown selectivity towards fluoride over acetate and justified the solid state binding stoichiometry.
 | ||
| Fig. 2 (a) Partial 1H-NMR (300 MHz) spectral changes of L in DMSO-d6 with added F− in DMSO-d6 (298 K), [L] = 10.05 mM. The respective ratio of concentrations are [F−]/[L]: (i) 0, (ii) 0.20, (iii) 0.46, (iv) 0.70, (v) 0.93, (vi) 1.16, (vii) 1.39, (viii) 1.62, (ix) 1.85, (x) 2.20, (xi) 2.55, (xii) 2.90. (b) Job's plot for L with F− in DMSO-d6 which shows a 1:2 stoichiometry. (c) Representative ITC titration profile of L (0.745 mM) with TBAAcO (14.45 mM) in DMSO at 298 K; thermodynamic parameters associated with this titration are, K1 = 4.17 × 103 ± 1.1 × 102 M−1, ΔH1 = −1795 ± 18.7 cal mol−1, ΔS1 = 10.5 cal mol−1 deg−1, K2 = 2.34 × 102 ± 8.3 M−1, ΔH2 = −2360 ± 84.5 cal mol−1, ΔS2 = 2.92 cal mol−1 deg−1, Chi2/DOF = 200.4. | ||
In conclusion, we have shown binding of anions and hydrated anions of different dimensionalities to a hexa-amide receptor with different structural arrangements (conformers A, B, C) in single crystal X-ray structural studies. Anion directed conformational diversity is observed in case of L, where we have observed the A conformer both in free ligand state and with acetate, B conformer with nitrate and thermodynamically highly unfavorable C conformer with fluoride through the isolation of discrete [F4(H2O)6]4− cluster. The solution state 1H-NMR titration and ITC studies have shown 1:2 binding stoichiometry with fluoride and acetate with fluoride selectivity. DFT calculations for each of the studied complexes have shown binding energies are sensitive to particular conformation (i.e., aaaaaa vs. aaabbb) as well as the electronegativity of the enclosed anion.
Acknowledgements
P. G. gratefully acknowledges the Department of Science and Technology (DST), New Delhi, India for financial support through Swarnajayanti Fellowship. S. C. and R. D. would like to acknowledge IACS, Kolkata, India for fellowship. B.M.W. acknowledges the National Science Foundation for supercomputing resources through the Extreme Science and Engineering Discovery Environment (XSEDE), Project no. TG-DMR140054.Notes and references
- (a) I. Ravikumar and P. Ghosh, Chem. Commun., 2010, 46, 1082–1084 RSC; (b) P. Bose, I. Ravikumar and P. Ghosh, Inorg. Chem., 2011, 50, 10693–10702 CrossRef CAS PubMed; (c) M. Li, B. Wu, C. Jia, X. Huang, Q. Zhao, S. Shao and X.-J. Yang, Chem.–Eur. J., 2011, 17, 2272–2280 CrossRef CAS PubMed; (d) N. Busschaert, M. Wenzel, M. E. Light, P.-I. Hernández, R.-P. Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed; (e) S. K. Dey, R. Chutia and G. Das, Inorg. Chem., 2012, 51, 1727–1738 CrossRef CAS PubMed; (f) B. Akhuli, I. Ravikumar and P. Ghosh, Chem. Sci., 2012, 3, 1522–1530 RSC; (g) P. Bose, R. Dutta, S. Santra, B. Chowdhury and P. Ghosh, Eur. J. Inorg. Chem., 2012, 5791–5801 CrossRef CAS; (h) M. A. Saeed, A. Pramanik, B. M. Wong, S. A. Haque, D. R. Powell, D. K. Chand and M. A. Hossain, Chem. Commun., 2012, 48, 8631–8633 RSC; (i) A. Pramanik, D. R. Powell, B. M. Wong and M. A. Hossain, Inorg. Chem., 2012, 51, 4274–4284 CrossRef CAS PubMed.
- (a) M. Arunachalam and P. Ghosh, Chem. Commun., 2009, 5389–5391 RSC; (b) P. Mateus, R. Delgado, P. Brandão and V. Fèlix, J. Org. Chem., 2009, 74, 8638–8646 CrossRef CAS PubMed; (c) M. Arunachalam and P. Ghosh, Inorg. Chem., 2010, 49, 943–995 CrossRef CAS PubMed; (d) P. Mateus, R. Delgado, P. Brandão and V. Fèlix, J. Org. Chem., 2012, 77, 4611–4621 CrossRef CAS PubMed; (e) N. L. Bill, D.-S. Kim, S. K. Kim, J. S. Park, V. M. Lynch, N. J. Young, B. P. Hay, Y. Yang, E. V. Anslyn and J. L. Sessler, Supramol. Chem., 2012, 24, 72–76 CrossRef CAS; (f) S. Chakraborty, R. Dutta, M. Arunachalam and P. Ghosh, Dalton Trans., 2014, 43, 2061–2068 RSC.
- (a) F. Hettche and R. W. Hoffmann, New J. Chem., 2003, 27, 172–177 RSC; (b) A. Frontera, F. Saczewski, M. Gdaniec, E. Dziemidowicz-Borys, A. Kurland, P. M. Deyà, D. Quiñonero and C. Garau, Chem.–Eur. J., 2005, 11, 6560–6567 CrossRef CAS PubMed; (c) I. Ravikumar and P. Ghosh, Chem. Commun., 2010, 46, 6741–6743 RSC; (d) R. Dutta and P. Ghosh, Eur. J. Inorg. Chem., 2013, 2673–2681 CrossRef CAS.
- (a) W. E. Allen, P. A. Gale, C. T. Brown, V. M. Lynch and J. L. Sessler, J. Am. Chem. Soc., 1996, 118, 12471–12472 CrossRef CAS; (b) G. Cafeo, F. H. Kohnke, G. L. L. Torre, A. J. P. White and D. J. Williams, Chem. Commun., 2000, 1207–1208 RSC; (c) A. Aydogan, D. J. Coady, S. K. Kim, A. Akar, C. W. Bielawski, M. Marquez and J. L. Sessler, Angew. Chem., Int. Ed., 2008, 47, 9648–9652 CrossRef CAS PubMed; (d) M. Ménand and I. Jabin, Org. Lett., 2009, 11, 673–676 CrossRef PubMed; (e) G. Cafeo, H. M. Colquhoun, A. Cuzzola, M. F. Gattuso, H. Kohnke, L. A. Valenti and J. P. White, J. Org. Chem., 2010, 75, 6263–6266 CrossRef CAS PubMed; (f) S. K. Kim, J. L. Sessler, D. E. Gross, C.-H. Lee, J. S. Kim, V. M. Lynch, L. H. Delmau and B. P. Hay, J. Am. Chem. Soc., 2010, 132, 5827–5836 CrossRef CAS PubMed.
- (a) Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. Garcia-España, Wiley-VCH, New York, 1997 Search PubMed; (b) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS; (c) K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS PubMed; (d) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry: Monographs in Supramolecular Chemistry, RSC Publishing, Cambridge, UK, 2006 Search PubMed; (e) S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 7882–7894 CrossRef CAS PubMed; (f) E. Garcia-España, P. Díaz, J. M. Llinares and A. Bianchi, Coord. Chem. Rev., 2006, 250, 2952–2986 CrossRef PubMed; (g) C. A. Schalley, Analytical Methods in Supramolecular Chemistry, Wiley-VCH, Weinheim, 2007 Search PubMed; (h) E. V. Anslyn, J. Org. Chem., 2007, 72, 687–699 CrossRef CAS PubMed; (i) P. A. Gale, S. E. García-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC; (j) S. Merchant and D. Asthagiri, J. Chem. Phys., 2009, 130, 195102–195111 CrossRef PubMed; (k) S. Kubik, Chem. Soc. Rev., 2009, 38, 585–605 RSC; (l) P. Ballester, Chem. Soc. Rev., 2010, 39, 3810–3830 RSC; (m) P. Ballester, Chem. Soc. Rev., 2010, 39, 3810–3830 RSC; (n) M. Arunachalam and P. Ghosh, Chem. Commun., 2011, 47, 8477–8492 RSC; (o) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed; (p) Anion Coordination Chemistry, ed. K. Bowman-James, A. Bianchi and E. Garcia-España, Wiley-VCH, New York, 2012 Search PubMed; (q) M. Cametti and K. Rissanen, Chem. Soc. Rev., 2013, 42, 2016–2038 RSC.
- F. Vögtle and E. Weber, Angew. Chem., Int. Ed. Engl., 1974, 13, 814–816 CrossRef.
- J. V. Gavette, A. L. Sargent and W. E. Allen, J. Org. Chem., 2008, 73, 3582–3584 CrossRef CAS PubMed.
- (a) M. Arunachalam and P. Ghosh, Org. Lett., 2010, 12, 328–331 CrossRef CAS PubMed; (b) M. Arunachalam and P. Ghosh, Chem. Commun., 2011, 47, 6269–6271 RSC.
- (a) M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809–2829 RSC; (b) M. A. Hossain, M. A. Saeed, A. Pramanik, B. M. Wong, S. A. Haque and D. R. Powell, J. Am. Chem. Soc., 2012, 134, 11892–11895 CrossRef CAS PubMed; (c) Q.-Q. Wang, V. W. Day and K. Bowman-james, J. Am. Chem. Soc., 2013, 135, 392–399 CrossRef CAS PubMed.
- (a) D. Das and L. J. Barbour, J. Am. Chem. Soc., 2008, 130, 14032–14033 CrossRef CAS PubMed; (b) D. Das and L. J. Barbour, Chem. Commun., 2008, 5110–5112 RSC; (c) D. Das and L. J. Barbour, Cryst. Growth Des., 2009, 9, 1599–1604 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterisation data of L & complex 1–3, Experimental section, H-bonding table of complexes, ITC titration profile, qualitative 1H-NMR spectra, 1H-NMR titration profiles, Job's plots and anion equivalents plot of receptors with CH3COO− & F−, DFT optimized structures of complexes 1–3 and Cartesian coordinates. CCDC 944132, 944135, 996874 and 996875. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra10795k |
| ‡ Color codes: carbon: yellow, oxygen: red, hydrogen: gray, nitrogen: cyan, fluorine: dark green. |
| This journal is © The Royal Society of Chemistry 2014 |