
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Support effect in the preparation of supported metal catalysts via microemulsion†
Riny Y.
Parapat
*,
Oey H. I.
Saputra
*,
Anton P.
Ang
,
Michael
Schwarze
and
Reinhard
Schomäcker
Technische Universität Berlin, Department of Chemistry, 10623 Berlin, Germany. E-mail: rinyyolandha@yahoo.de; schomaecker@tu-berlin.de
First published on 2nd October 2014
Abstract
It is well known that the activities of supported metal catalysts are strongly dependent upon the size, shape and dispersion of the nanoparticles on the support material. There are several techniques which can be implemented in order to produce such catalysts, e.g. wet impregnation, however the deposition of nanoparticles (NPs) on the support material without agglomeration still proves a challenge. This is particularly significant when attempting to maintain the size and shape of the particles during the deposition process. We have introduced a new method to deposit metal NPs, namely thermo-destabilization of microemulsions (please see J. Mater. Chem., 2012, 22, 11605–11614 and Nanoscale, 2013, 5, 796–805), in which the NPs are formed prior the deposition process. This method is an ingenious approach to control the dispersion of NPs on the support material and depositing NPs evenly with a narrow size distribution. In this paper we expound the important role of the surface charges of NPs and the support material, as indicated by zeta potentials, on the metal dispersion, and how they affect the catalytic activity. We also investigate the influence of other parameters such as the pore size and the pre-calcination of the support on the catalytic activities of the resulting supported metal catalysts.
1. Introduction
Supported metal catalysts have more advantages in many respects than unsupported metal catalysts.1,2 The primary aim of depositing active metal nanoparticles (NPs) onto a support material is to obtain the catalyst in a highly dispersed form. The catalytic performance is not only affected by the size and shape of the deposited NPs, but is also significantly affected by their dispersion. Improving the dispersion of a metal catalyst on the support material generally increases the activity.3–7 This is particularly important with regard to precious metal catalysts, because it allows for more effective and economic usage of the metal than can be achieved in bulk-metal systems.8 Particle dispersions are strongly affected by the nature and surface structure of the support material.4 Another reason for using a support for the metal catalyst is to improve the catalyst's stability. The catalytic stability is also dependent upon the nature of the support on which the metal is dispersed and on the interaction between the metallic phase and the support, the latter being the controlling parameter of the sintering process.9 In addition, the support facilitates metal recovery and provides a greater resistance to poisoning. In contrast, unsupported metal is used less efficiently than supported metal and recovery losses are likely to be higher.1A common technique used to produce supported metal catalysts is carried out by impregnating the support with a solution containing a dissolved metal salt precursor followed by reduction and calcination. The principle of this deposition method is to adsorb a metal salt onto the support surface and then to reduce the salt to the metal at high temperatures under an atmosphere of H2 or Ar.10 Another way is to first produce the metal NPs and subsequently deposit them onto the support material.4,11–17 The latter has more advantages than the former, because the size and shape of the nanoparticles are more controllable (they can be designed first and posses a narrow crystallite distribution) in comparison to those obtained through the traditional impregnation, co-precipitation, and precipitation–deposition methods. This way can also be applied to deposit the subnanometer metal clusters onto the support material.18
Particle dispersion is highly influenced by the method of catalyst synthesis. The preparation of support material has a great effect on dispersion.19 Theoretically, the electric surface charge of either the support or the metal NPs affects their interaction during the deposition process. Bianchi et al. found that the surface charges of particles influence the final patterns of the particles.20 They stated that the overall particle charge affects the ratio of directional attractive and repulsive contributions to the effective interactions. By the same principle, the surface charges of particles and the support material both influence the dispersion pattern of particles on the support. The greater the contrast between the surface charge of the metal NPs and the support, the stronger the resulting bond will be. It will also be more difficult for the particles to migrate or loosen from the support, and thus their tendency to agglomerate will be reduced.
This paper mainly discusses the deposition of different metal NPs onto the support surface instead of impregnation. We synthesized metal NPs first by using a water in oil microemulsion system and subsequently deposit them onto the support. In this method, the inner core of the water droplet is considered as a nanoreactor to prepare and design the catalyst NPs. Boutonnet et al.21,22 has done pioneering work in this field. They prepared platinum catalysts by depositing on alumina monodispersed particles of platinum produced in reversed micellar solution. In our work, the NPs were transferred onto the supports by a facile method, namely themo-destabilization of microemulsions. During this procedure a thin water layer is formed at the surface of the support material from the water that is released from the droplets. We discovered that by simply contacting the opening droplets containing NPs with the support, spontaneous deposition onto the support could occur. However, as mentioned above, the surface charges of NPs and the support influence the particle dispersion. Application of the same synthesis procedure to deposit various metals on the same kind of support material may lead to catalysts with different features. Because metals have different surface charges, when they are deposited onto the same support material the dispersions and loading of obtained supported metals will vary. Although many have described the influence of the support material on the metal loading and the catalyst performance,4,23–27 there is no particular explanation yet concerning the influence of the surface charges of the support and the metal NPs upon the metal loading, dispersion, and activity.
In our previous paper,12 we observed that the dispersion of metal NPs on the support also depends on the properties of the support. In the case of Pt deposition, we found that the obtained activities of Pt NPs (prepared using the same conditions) supported on SBA-15 and Al2O3 are extremely different. It is most likely that not only the high surface area of SBA-15 (800 m2 g−1), which favors the releasing of particles, but also the structure of the support is responsible for the activity. The well ordered channel structure of SBA-15 allows a good dispersion and can also hinder metal NPs from sintering, thus promoting good contact between active sites and reactants.
To obtain a well dispersed supported catalyst, it is necessary to promote a homogeneous distribution when transferring the NPs onto the support. In addition, the NPs have to adhere strongly to the support in order to minimize the sintering of the particles at high temperatures.28,29 In our method a proper mixing of the microemulsions, which contain metal NPs together with the support particles will increase the possibility of obtaining a homogeneous distribution of the particles on the support. During the deposition process, the adhesion of the metal NPs to the support material is dependent on the electrostatic interaction between the NPs and the support surface. Two outcomes might happen. If the interactions are too weak, the metal NPs would be loosely bound to the surface. This may lead to migration of the particles, especially during the calcination step, and possibly result in the formation of larger agglomerates. However, a weak interaction may also lead to the metal particles penetrating further into the support pore system before attaching to its surface during the deposition step. When the interactions are relatively strong, the NPs are not likely to migrate during the calcination. This may lead to less agglomeration which results in higher catalytic activity. How these two phenomena affect the dispersion of the NPs during the deposition will be verified in this study. In this paper we also elucidate the thermo-destabilization of microemulsions in the preparation of different supported metal catalysts.
2. Experimental section
2.1 Chemicals
As metal precursors in the preparation of different metal nanoparticles via the microemulsion method: hexachloroplatinic acid hydrate (H2PtCl6, 99.9% purity, Sigma-Aldrich), potassium tetrachloro-platinate(II) (K2PtCl4, 99.9% purity, Sigma-Aldrich), silver nitrate (AgNO3, 99.9% purity, Sigma-Aldrich), palladium chloride (PdCl2, 99.9% purity, Sigma-Aldrich) and ruthenium chloride (RuCl3, 99.9% purity, Sigma-Aldrich) were used. Hydrazine monohydrate (N2H4, 98% purity, Sigma-Aldrich) and L-(+)-ascorbic acid (AA; >99% purity, Alfa Aesar) were used as reducing agents.2-[4-(2,4,4-trimethylpentan-2-yl)phenoxy]ethanol (trade name: Triton X-100, ∼100% purity, Sigma-Aldrich) was used as the surfactant 1-pentanol (≥98% purity, Carl-Roth) was used as the co-surfactant and cyclohexane (≥99.5% purity, Carl-Roth) was used as the oil. As supports materials in the deposition process basic, neutral and acidic alumina oxide ((base, neutral and acid)-Al2O3, Sigma-Aldrich), silica (SiO2, Sigma-Aldrich), α-alumina oxide (α-Al2O3, Alfa Aesar), dioxosilane (trade name: Sipernat 310, Evonik) and SBA-15 as well as MCM-41 synthesized according to the method reported by Zhao et al.30 Acetone (≥99.8% purity, Carl Roth) was used to wash the catalysts after the synthesis.
For catalytic testing in hydrogenation reactions α-methyl styrene (AMS, 99% purity, Sigma-Aldrich) and methyl crotonate (CME, 99% purity, Sigma-Aldrich).
The substrates that are used for catalytic testing in hydrogenation reactions are α-methyl styrene (99% purity, Sigma-Aldrich) and methyl crotonate (99% purity, Sigma-Aldrich). As the solvent, methanol (≥99.9% purity, Carl-Roth) was used. All chemicals were used as received.
2.2 Characterization and analysis methods
The shape and size of the metal NPs were investigated with transmission electron microscopy (TEM) using a FEI Titan 80-300 (sub-angstrom resolution, which is able to investigate at an atomic scale), a FEI Tecnai G2 S-Twin TEM, and a Philips CM200/FEG high-resolution TEM (HRTEM) operated at 200 kV. The microscopes are equipped with an energy dispersive X-ray detector (EDX). The metal content of the supported catalysts was analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES) using an Element 2 (Varian), sample gas 0.863 L min−1; plasma power 1350 W. The effective surface charges (zeta potential) for the metal NPs and support materials were measured using a Zen 3600 Zetasizer (Malvern Instruments, Worcestershire, UK). Samples were measured at 25 °C. The specific surface areas of the catalysts were measured by implementing the BET method using N2 adsorption at liquid N2 temperature in a Micromeritics Gemini III 237 Volumetric Surface Analyzer. The sample was outgassed at 200 °C for 1 h to desorb any impurities or moisture from its surface.2.3 Synthesis of supported metal nanoparticles by thermo-destabilization of microemulsions
The synthesis process as well as the reaction conditions are described in detail in our previous papers.11,12 The key principle of this synthesis method is to utilize a temperature change to destabilize the microemulsions so that the metal NPs which are formed inside of the droplets by reduction of the metal precursor can be released and attached onto the support material. In addition, a composition of the microemulsion is needed, which is one phase at room temperature. Therefore, it is necessary to establish the phase behavior of the microemulsion system prior to the nanoparticle formations so that the corresponding destabilization temperature can be identified. The phase behaviour of microemulsions systems are shown in detail in our previous paper.11 The key parameters are the oil fraction (α), the surfactant fraction (γ), the co-surfactant ratio (δ), and the water to surfactant molar ratio (ω). Their definitions are given in eqn (1)–(4)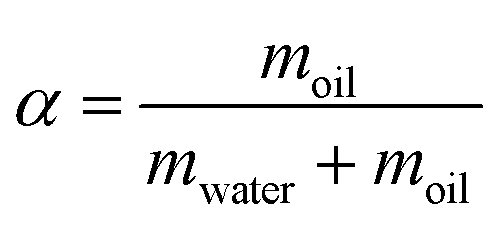 | (1) |
 | (2) |
 | (3) |
 | (4) |
After determining the destabilization temperature of the microemulsions, the synthesis of supported NPs was carried by mixing two microemulsions having a volume of 50 ml each, one containing the reducing agent and one containing the dissolved metal precursor. Table 1 shows the concentration of metal salts and reducing agents (without the variation of concentrations) with different compositions that are used in this work. The activities of resulting catalysts were tested in hydrogenation reaction which is described in ESI.†
| Microemulsion component | Microemulsion I | Microemulsion II | Mass fraction | ||
|---|---|---|---|---|---|
| α 1 = 0.92 γ1 = 0.3 (ω1 = 12.96) | α 2 = 0.5 γ2 = 0.7 (ω2 = 14.88) | α 3 = 0.75 γ3 = 0.4 (ω3 = 26.96) | |||
| Water phase | H2PtCl6 (0.31 mM) | N2H4 (2.15 mM) | 5.6 | 15 | 15 |
| H2PtCl6 (0.31 mM) | AA (15.38 mM) | ||||
| AgNO3 (0.56 mM) | N2H4 (5.60 mM) | ||||
| PdCl2 (0.56 mM) | AA (28.19 mM) | ||||
| RuCl3 (0.59 mM) | N2H4 (5.90 mM) | ||||
| Oil phase | Cyclohexane | Cyclohexane | 64.4 | 15 | 45 |
| Co-surfactant | Pentanol | Pentanol | 15 | 35 | 20 |
| Surfactant | Triton X-100 | Triton X-100 | 15 | 35 | 20 |
3. Result and discussion
3.1 Particle deposition with the thermo-destabilization of microemulsions
In the thermo-destabilization of microemulsions, the deposition process starts by destabilizing (breaking) the water droplets by means of increasing the temperature of the microemulsions and subsequently releasing the metal NPs. The principle and procedure of particle deposition via the thermo-destabilization of microemulsions which is based on the phase diagram (Fig. 1) has been explained in our previous paper.11 | ||
| Fig. 1 Illustration of metal NPs deposition on a support material via the thermo-destabilization of microemulsions. The inset is a TEM image of the deposited metal NP. The metal NP which is formed by mixing the one phase microemulsions containing metal precursor (M+) and reduction agent (Red) is deposited onto the support by increasing the temperature above the phase boundary of the microemulsions. | ||
Due to the aqueous core of the microemulsion droplets and the water layer that is formed at the surface of the support during the deposition process, we consider solid–water–solid interaction which result from a combination of van der Waals and electrostatic interactions. To facilitate the interaction between the metal NPs and the support, mixing with an appropriate rate (700 rpm) was applied during the deposition process. In our experiment, we use anionic surfactant, which exhibits weak hydrogen bonding to the support material. It can therefore be assumed that the surfactant has no effect on the deposition of the metal to the support. It is also important to note that we found already in our previous studies11,12 that the particle size analysis before and after deposition shows no increase in size. Therefore, in our case, Ostwald ripening or other growth mechanisms of the particles can be excluded.
3.2 Effect of the zeta potential on the deposition yield
It is generally known that particles with an identical charge like anions will tend to repel each other and those with opposite charge like cations and anions will attract each other. The metal NPs that are produced by nuclei growth have a certain charge on the surface. A metal nanoparticle is surrounded by a layer which contains ions with the opposite charge. The ions which are further away from the nanoparticle create a diffuse layer which is more loosely bound (Fig. 2). In this diffuse layer, there is a plane of a speculative boundary which is known as the surface of the hydrodynamic shear or the slipping plane.31 The potential at this boundary is known as the zeta potential. The value of the zeta potential is affected by both the nature of the particle's surface and the dispersant.32 The zeta potential of metal nanoparticles and support materials need to be measured in order to observe the effect of the surface charges of both materials on the deposition yield and the activity of the resulting catalysts. Table 2 presents the experimentally measured zeta potentials of the support materials and metal nanoparticles that are used in this application.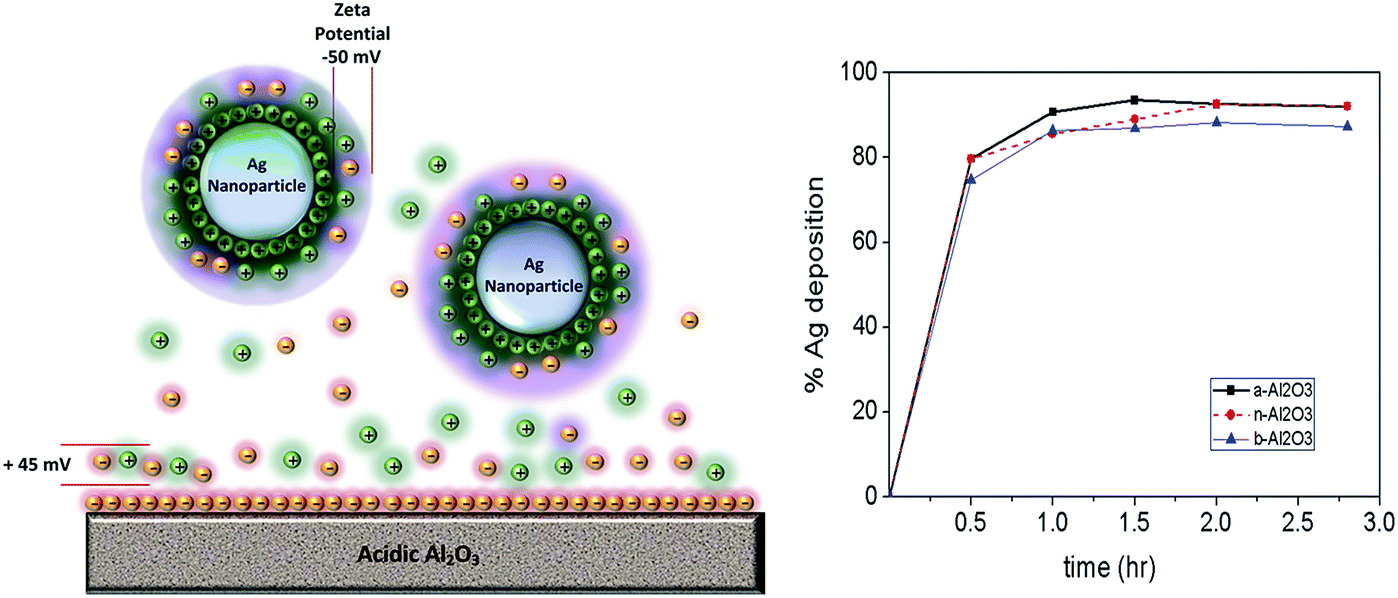 | ||
| Fig. 2 Left: schematic illustration of the situation around the support material when Ag NPs are released from the water droplets and then attached onto acidic alumina. Right: deposition yield of Ag NPs on acidic, basic, and neutral alumina based on UV vis spectrometer measurement. | ||
Fig. 2 shows the deposition yields of the Ag NPs on three alumina supports with differing acidities. Here we can see clearly the effect of zeta potential on the successful deposition (nearly 100%) of Ag NPs on alumina supports, although a slight difference with regard to the yield percentage is observed. As we can see from Table 2, the zeta potential of the Ag NPs is around −50 mV, whereas those of the acidic, neutral, and basic Al2O3 supports, were +45 mV, +20 mV, and +10 mV, respectively. Therefore, the deposition yield of the Ag NPs on acidic Al2O3 is slightly higher than the other Al2O3 supports. This indicates that the highly negative charge of Ag NPs is still attracted to slightly positive charged supports such as basic alumina. These findings indicate that aluminum oxide is a very suitable catalyst support for Ag NPs.
Fig. 3 shows that the deposition yield of Ag NPs (−50 mV) on the SiO2-Sipernat 310 is higher than SiO2-pellet although they have the same charge (∼ −20 mV). We attribute this to the size of Sipernat 310 (7.5 μm) which is much smaller than SiO2-pellet (3000 μm), that promotes a good contact with the Ag NPs. It is interesting that although both Ag NPs and SiO2 particles have negative surface charges, the deposition process can still take place. We attribute this to the attractive interaction, i.e. van der Waals interaction, in which the nanoparticle adsorbs like-charged ions onto its surface as also described by Walker et al.33
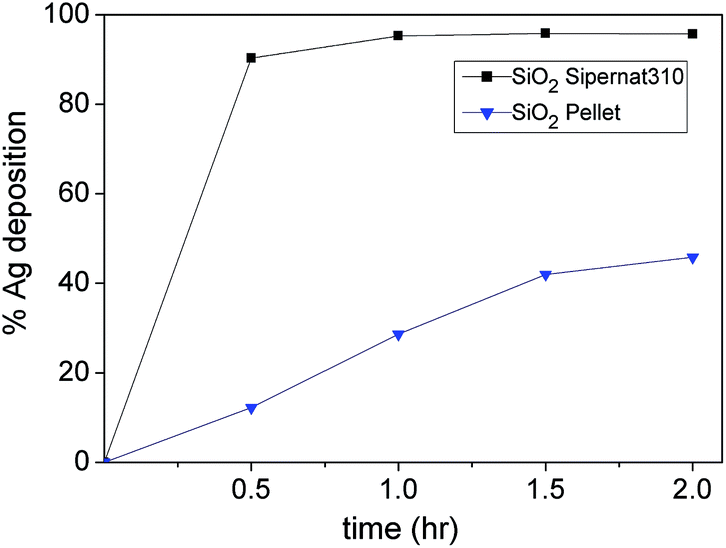 | ||
| Fig. 3 Deposition yield of Ag NPs on different silica supports based on UV vis spectrometer measurement. | ||
Fig. 4 (left) shows the deposition yields of different metals (Ru, Pd, Pt, Ag) on basic alumina which are prepared in the same conditions. Here we can see that the more contrasting the zeta potential between the metal and the support, the higher the deposition yield of the support material. In order to increase the percentage loading of the metal on the support material, the amount of the metal in the microemulsions is increased while the amount of the support is fixed. Fig. 4 (right) illustrates the profiles of percentage loading of Ag, Pd, Ru and Pt on 1000 mg of basic alumina versus the amount of metal in the microemulsions. Among other metals, Ag nanoparticles show the highest slope followed by Pd, Ru and Pt, respectively. In this case, the slope signifies the driving force for deposition which is the metal's affinity to the support material. This confirms the fact that Ag has the most contrasting zeta potential (−50 mV) to basic alumina (+10 mV) in comparison to Pt, Ru and Pd which are −25, +57 and +24 mV respectively. Here we see again that although Pd and Ru have positive charges as the alumina support the deposition still can take place due to the van der Waals interaction, but with low deposition yields.
 | ||
| Fig. 4 Left: deposition yields of different metals on basic Al2O3 with a thermo-destabilization method. The amount of metal precursor prepared in each microemulsion system is 6 mg. Right: loading of different metal catalysts on basic alumina as a function of the amount (mg) of corresponding metal in the precursor solutions. | ||
To see further the effect of surface charge on the deposition process, Ru and Pd NPs were deposited on the same provided surface area of different supports (300 m2 g−1). As depicted in Fig. 5, the loadings of Ru and Pd NPs are higher on SBA-15 and MCM-41 compared to other supports due to the contrast of the zeta potentials. The highest loadings of Ru and Pd NPs on MCM-41 are likely due to support structure that promotes a good dispersion as shown in SEM images of Fig. 8.
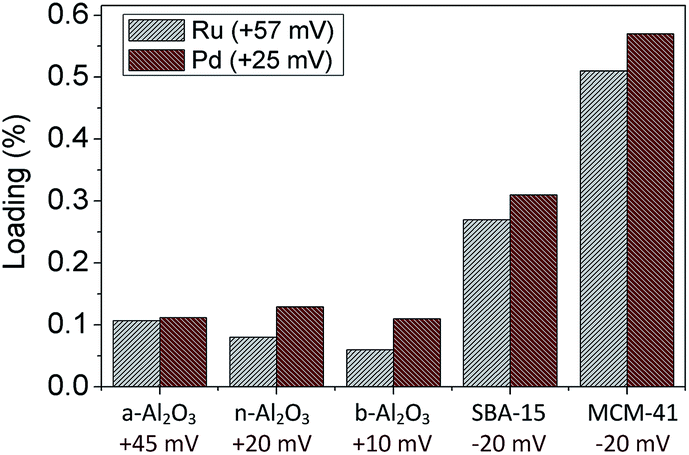 | ||
| Fig. 5 Loading of supported Ru and Pd catalysts on 300 m2 g−1 support materials. The amount of metal in the precursor solutions is 6 mg. | ||
 | ||
| Fig. 6 Activities of the Pt nanodendrites deposited on acidic Al2O3 and SBA-prepared in different molar ratios of Pt to AA and the corresponding TEM images. The larger particles produced by a higher molar ratio of Pt to AA cannot permeate the pores of SBA-15. The lower loadings of Pt@SBA-15 are caused by the same particles charges of Pt and SBA-15. | ||
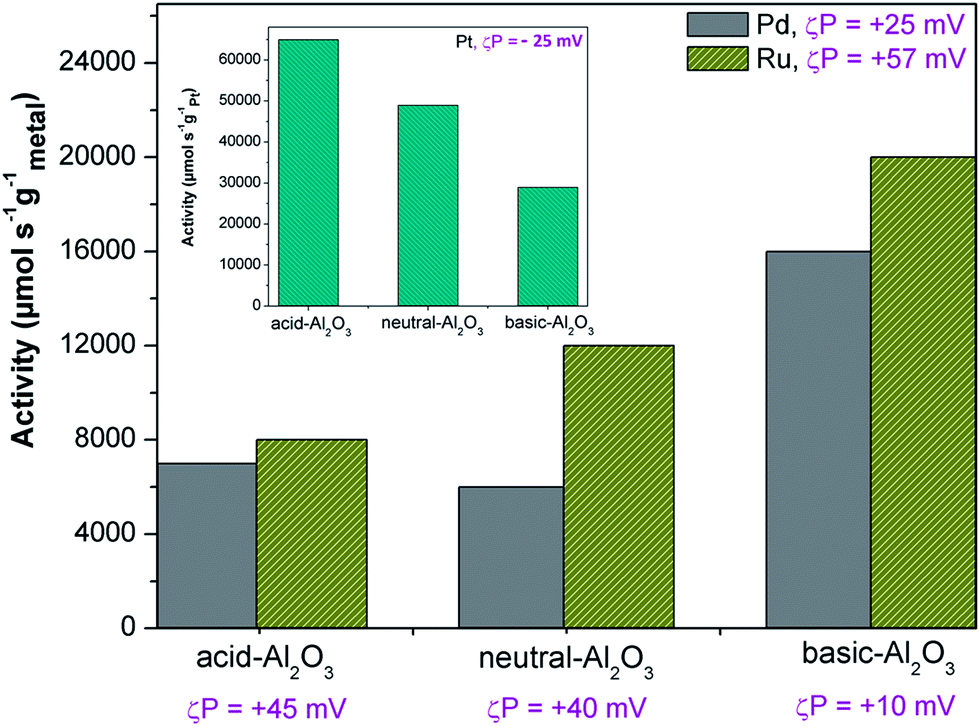 | ||
| Fig. 7 Activities of Pd and Ru catalysts supported on different acidity of Al2O3 in hydrogenation of methyl crotonate. The inset is activities of Pt catalyst supported on different acidity of Al2O3 in hydrogenation of α-methyl styrene. The opposite trend of activities between Pt catalyst and Pd and Ru catalysts is because of the different value of zeta potential. | ||
 | ||
| Fig. 8 Upper left: activities of supported Pt catalysts in hydrogenation of α-methyl styrene. The Pt catalyst was reduced with AA on deposited on acidic (A), neutral (B), basic (C) alumina supports. Upper right: degrees of dispersion of the corresponding catalysts with the Delaunay network method.34 Bottom: bright and dark field TEM images of the corresponding catalysts. | ||
3.3 Effect of the support nature on the catalytic activity
To study the influence of the support nature on the catalytic activity, we deposited Pt nanodendrites, which were prepared by the same system (using AA as the reductant), on different supports (SBA-15 and acidic-Al2O3). Here we found that the activities of the resulting catalysts are extremely different as indicated in Fig. 6. When Pt nanodendrites are deposited on acidic-Al2O3, although the sizes of particles that are produced by a Pt to AA ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 (4.6 ± 1.2 nm) are smaller than 1:50 (7.7 ± 2.6 nm), the activity of supported larger particles is higher. However, when the larger particles produced from [Pt]:[AA] of 1:50 are deposited on the SBA-15, the activity of resulting catalyst is decreased. This happens because the larger particles cannot penetrate the pores and have a tendency to agglomerate. The smaller particles (less than 6 nm) produced from [Pt]:[AA] of 1:25, in contrast, can penetrate the pores of the SBA-15 which could result in a good dispersion. Therefore, in the case of deposition on SBA-15, increasing the particle size leads to decreased activity. In this case, the support pores affect the particle dispersion and the metal particle size. The smaller number of particles that can be deposited onto SBA-15 (low loading), in comparison to the alumina support, is due to the similar charges of the Pt particles and support, which are −25 mV and −20 mV, respectively, whereas alumina is +45 mV.
:25 (4.6 ± 1.2 nm) are smaller than 1:50 (7.7 ± 2.6 nm), the activity of supported larger particles is higher. However, when the larger particles produced from [Pt]:[AA] of 1:50 are deposited on the SBA-15, the activity of resulting catalyst is decreased. This happens because the larger particles cannot penetrate the pores and have a tendency to agglomerate. The smaller particles (less than 6 nm) produced from [Pt]:[AA] of 1:25, in contrast, can penetrate the pores of the SBA-15 which could result in a good dispersion. Therefore, in the case of deposition on SBA-15, increasing the particle size leads to decreased activity. In this case, the support pores affect the particle dispersion and the metal particle size. The smaller number of particles that can be deposited onto SBA-15 (low loading), in comparison to the alumina support, is due to the similar charges of the Pt particles and support, which are −25 mV and −20 mV, respectively, whereas alumina is +45 mV.
In the synthesis of Pt@C3N4, which can be used as photocatalyst for sacrificial water reduction, only very few Pt dendrites can be deposited (less than 0.1 wt%). We attribute this to the fact that the zeta potentials of C3N4 (at pH 4) and Pt dendrites are almost the same, which are −20 and −24 mV respectively, and that the Pt dendrites which have a size of more than 5 nm have difficulty penetrating the support C3N4 which has the pore size of 2–4 nm.
It is observed that the zeta potential indirectly affects the activity of the produced catalyst, as shown in Fig. 7. The activities of supported Ru catalysts are higher than Pd catalysts most probably because Ru catalysts have a better dispersion than Pd catalysts. Ru is more positively charged (+57 mV) than Pd (+25 mV), whereas the zeta potential of acidic, neutral, and basic Al2O3 are +45, +40 and +10 mV, respectively. The more positively charged support particles are more likely to interact with the less positively charged metal particles which results in better dispersion. Therefore, in this case, the dispersion of Ru is better on basic Al2O3 followed by neutral Al2O3 and acidic Al2O3. This result is opposite of the activity of Pt particles in the hydrogenation of α-methyl styrene (see the inset).
According to Table 2, the zeta potential of the Pt is about −25 mV whereas the zeta potentials of acidic, neutral, and basic Al2O3 are +45, +40, and +10 mV, respectively. The dispersion of Pt particles seems to be better on acidic Al2O3 followed by neutral Al2O3 and basic Al2O3 which results in an activity decrease of Pt catalysts with the support material ranking from acidic to basic Al2O3. To confirm the visual observations, we quantify the dispersion by using the Delaunay network method based on the TEM images in Fig. 8. The details of the Delaunay network method are described in the ESI.† As we can see in Fig. 8 (upper right), according to this method, the dispersions of Pt particles on the acidic, neutral, and basic alumina are in the areas of good, random-like, and poor, respectively. These quantified results support the visual observations and certainly agree with the catalytic activities.
A relatively strong interaction between the Pt NPs and the acidic-Al2O3 makes it hard for the Pt NPs to migrate or loosen from the support during deposition. As a result, these Pt particles have least tendency to agglomerate, thus increasing the catalytic activity. A weaker electrostatic interaction between the Pt NPs and the basic-Al2O3 support, in contrast, seems to cause more agglomeration of Pt particles which results in a decrease of catalytic activity.
To confirm further the effect of the support type on the activity of supported catalysts, the supported Pd and Ru catalysts are investigated. As we can see in Fig. 9, the Pd and Ru catalysts have the highest activity in methyl crotonate hydrogenation when supported on MCM-41 followed by SBA-15 and Al2O3. The most probable reason for this account is the structure of the support. Fig. 9 shows clearly the structure of the supported Pd catalysts by the SEM images. The structure of MCM-41 seems to promote better dispersion of Pd catalyst compared to SBA-15 and Al2O3. In addition, if we compare the zeta potentials of MCM-41, SBA-15, and Al2O3 which are −20, −25, and +20 mV, respectively, with the zeta potentials of Pd and Ru, which are +25 and +57 mV, we can conclude that both metals are not well dispersed on alumina.
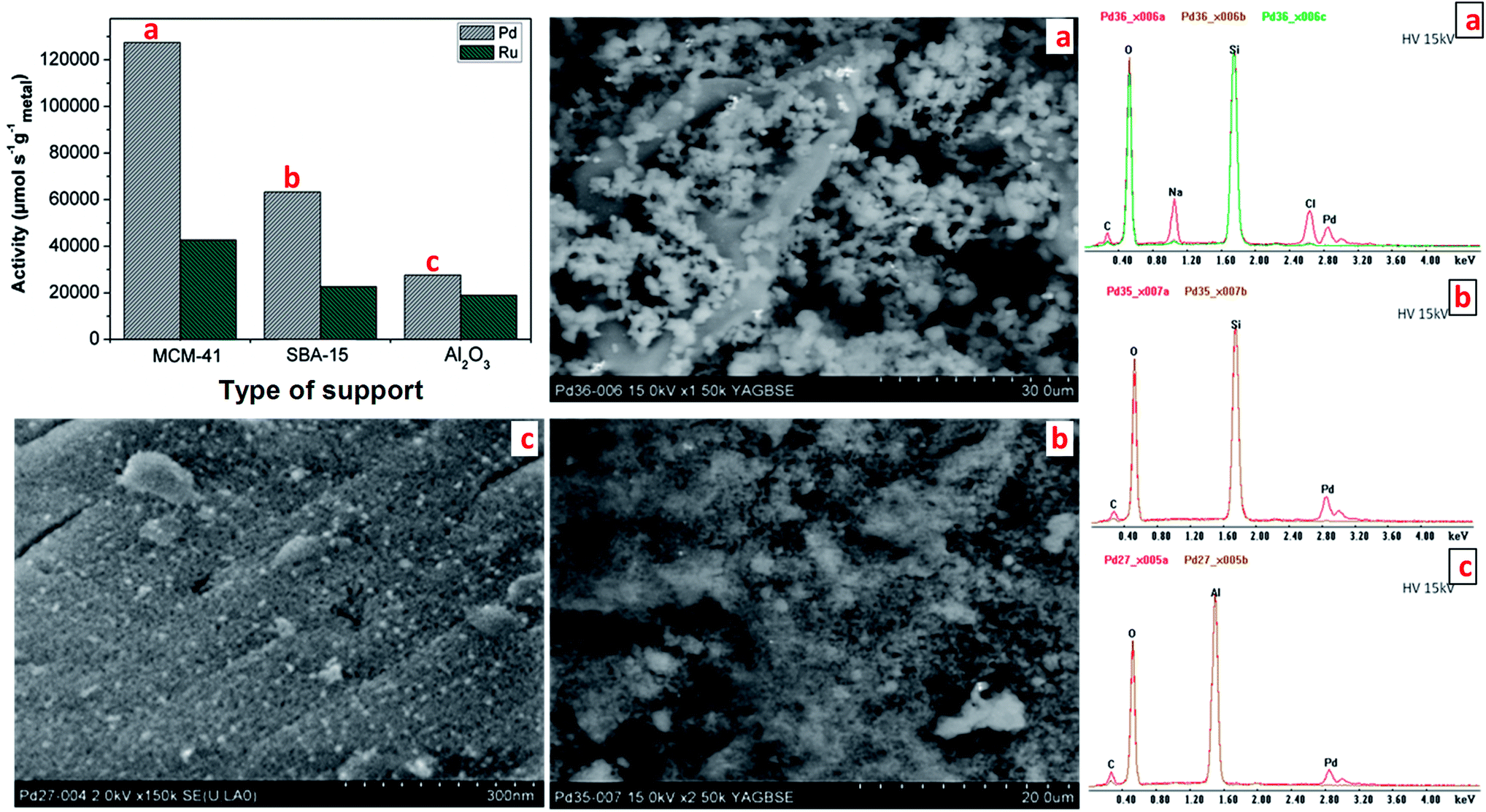 | ||
| Fig. 9 Activities of Pd and Ru NPs deposited on different supports in hydrogenation of methyl crotonate at 20 °C and 1.1 bar with the corresponding SEM images and EDX of supported Pd catalysts. | ||
3.5 Effect of pre-calcination of the support
Another possible method to improve the deposition yield and activity is to preheat the support material (pre-calcination). The support was preheated to 500 °C and held for 2 h with the intention of removing any moisture inside of the support pores so that the particles can get into the pores without barrier. Here, Pt and Pd nanoparticles were synthesized using ascorbic acid as the reductant and deposited on basic-Al2O3, either with or without pre-calcination. Interestingly, there is no significant difference between yields of deposition of both metals on the support, with and without pre-calcination, as shown in Table 3. However, in the case of activity, only the supported Pt catalysts show substantial difference, with and without pre-calcination of the support. This might be attributed to the metal dispersion on the support with the correlation to the value of the zeta potential of the involved materials.| Pretreatment of support (basic-Al2O3)a | Pd/basic-Al2O3b | Pt/basic-Al2O3c | ||
|---|---|---|---|---|
| Deposition yield (%) | Activityd (μmol s−1 gPd−1) | Deposition yield (%) | Activitye (μmol s−1 gPt−1) | |
| a ζ-potential of basic Al2O3 = +10 mV. b ζ-potential of Pd nanoparticles = +25 mV. c ζ-potential of Pt nanoparticles = −24 mV. d Tested in α-methyl styrene hydrogenation. e Tested in methyl crotonate hydrogenation. | ||||
| Without pre-calcination | 35.3 | 13000 |
36.8 | 6000 |
| Pre-calcination (500 °C, 2 h) | 35.7 | 9000 | 33.9 | 29000 |
Because the value of the zeta potential of Pd NPs (= +25 mV) is not so different to the basic-Al2O3 (= +10 mV), pre-calcination has no significant effect on the dispersion of NPs on the support. In fact, they both are positively charged materials. However, in the case of deposition of Pt NPs (= −25 mV) on basic-Al2O3, pre-calcination has a considerable effect on the NPs dispersion.
This may also indicate that the pre-calcination process causes the Al2O3 to lose its –OH group to such an extent that it becomes more acidic. Miller et al. reported that increasing the alumina pre-calcination temperature results in a decrease in the intensity of the hydroxyl bands, although a significant number of hydroxyl bands remain even after calcination at 700 °C. They found that in the alumina catalysts, the number of surface hydroxyl groups was varied by changing the support precalcination temperature. As the calcination temperature increased, the number of hydroxyl groups decreased as evidenced by infrared spectroscopy.35
The acidic Pt nanoparticles (zeta potential = −24 mV) are more stable before attaching to the acidic support which results in less agglomeration on the support, leading to higher activity. In the case of supported Pd catalysts, although there is not so much difference in the activity (Table 2), the alkalinity of Pd NPs (zeta potential = +25 mV) causes them to be less stable before attaching to the more acidic support. As a result, it produces Pd agglomeration on the support, leading to the lower activity. As can be seen in Fig. 10, the dispersion of the Pd NPs on basic alumina without precalcination is better than those on the precalcinated one. The few particles on the precalcinated support indicate that the dispersion is not uniform.
 | ||
| Fig. 10 Effect of precalcination of the support material before synthesis on the catalytic activity of Pd particles supported on basic alumina and the corresponding SEM images. | ||
The same deposition yields of both supported Pt and Pd catalysts with and without precalcination (Table 3) may lead to the conclusion that the loss of some OH groups of the support does not influence the adsorption of particles. Therefore, this phenomenon is not the same as that which happens in the deposition with impregnation technique, which generally shows noticeable effect of OH groups on either the deposition yield or the dispersion.36,37 Here we would also like to note that in our case, we do not expect any strong metal-support interaction (SMSI) effect because there is no partial reduction on the surface of the alumina and silica support which are used in the preparation. The occurrence of SMSI requires the reducibility of the support, whereas alumina (Al2O3) and silica (SiO2) are very resistant to reduction.
4. Conclusion
Supported Pt, Ag, Pd, and Ru catalysts have been prepared via thermo-destabilization of microemulsions. Although they were prepared by the same procedure, their features are different from one another. The zeta potentials of metal NPs and the support material play an important role in the dispersion of the particles. A relatively strong interaction between the metal particles and the support makes the metal particles steadily attach to the support during deposition, and therefore reduces the tendency to agglomerate which in turn leads to higher catalytic activity. A weaker electrostatic interaction between the Pt particles and the basic-Al2O3 support, in contrast, seems to cause more agglomeration of Pt particles which results in a decrease of catalytic activity. Therefore, to provide good particle dispersion, it is important to consider the zeta potentials of the metal particles and support materials when choosing the support material. Additionally, the pore size of the support must be bigger than the particle to promote a good dispersion, which results in higher activity. Pre-calcination of the support material has insignificant effect on the deposition yield and could decrease the catalytic activity due to poor dispersion.Acknowledgements
The authors are grateful to the Ministry of National Education of Indonesia (Dikti) for financial support. Thanks to Patrick Littlewood and Cody Nichol for helping in writing this paper, Dipl. Sören Selve for performing the TEM/HRTEM measurements and to Astrid Müller for assisting with the ICP measurements. This work is also a part of the DFG funded Cluster of Excellence “Unifying Concepts in Catalysis”.References
- P. Rylander, Catalytic Hydrogenation over Platinum Metals, Elsevier, 2012 Search PubMed.
- G. C. Bond, Platinum Metals Rev., 1975, 19, 126 Search PubMed.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014 10.1039/C3CS60395D.
- C.-J. Jia and F. Schüth, Phys. Chem. Chem. Phys., 2011, 13, 2457–2487 RSC.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- K.-J. Kim, Y.-J. You, M.-C. Chung, C.-S. Kang, K.-H. Chung, W.-J. Jeong, S.-W. Jeong and H.-G. Ahn, J. Nanosci. Nanotechnol., 2006, 6, 3589–3593 CrossRef CAS PubMed.
- N. Linares, A. M. Silvestre-Albero, E. Serrano, J. Silvestre-Albero and J. García-Martínez, Chem. Soc. Rev., 2014 10.1039/C3CS60435G.
- S. F. Chen, J. P. Li, K. Qian, W. P. Xu, Y. Lu, W. X. Huang and S. H. Yu, Nano Res., 2010, 3, 244–255 CrossRef CAS.
- P. Ferreira-Aparicio, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Appl. Catal., A, 1998, 170, 177–187 CrossRef CAS.
- G. G. Wildgoose, C. E. Banks and R. G. Compton, Small, 2006, 2, 182–193 CrossRef CAS PubMed.
- R. Y. Parapat, V. Parwoto, M. Schwarze, B. Zhang, D. S. Su and R. Schomäcker, J. Mater. Chem., 2012, 22, 11605 RSC.
- R. Y. Parapat, M. Wijaya, M. Schwarze, S. Selve, M. Willinger and R. Schomäcker, Nanoscale, 2013, 5, 796–805 RSC.
- S. Li, G. Liu, H. Lian, M. Jia, G. Zhao, D. Jiang and W. Zhang, Catal. Commun., 2008, 9, 1045–1049 CrossRef CAS PubMed.
- H.-S. Qian, M. Antonietti and S.-H. Yu, Adv. Funct. Mater., 2007, 17, 637–643 CrossRef CAS.
- D. C. Lee, D. K. Smith, A. T. Heitsch and B. A. Korgel, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2007, 103, 351 RSC.
- Y. Liu, J. Goebl and Y. Yin, Chem. Soc. Rev., 2013, 42, 2610–2653 RSC.
- O. B. Shawkataly, R. Jothiramalingam, F. Adam, T. Radhika, T. M. Tsao and M. K. Wang, Catal. Sci. Technol., 2012, 2, 538–546 Search PubMed.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- S. L. Suib, New and Future Developments in Catalysis: Catalysis by Nanoparticles, Newnes, 2013 Search PubMed.
- E. Bianchi, C. N. Likos and G. Kahl, ACS Nano, 2013, 7, 4657–4667 CrossRef CAS PubMed.
- M. Boutonnet, J. Kizling, P. Stenius and G. Maire, Colloids Surf., 1982, 5, 209–225 CrossRef CAS.
- M. Boutonnet, S. Lögdberg and E. Elm Svensson, Curr. Opin. Colloid Interface Sci., 2008, 13, 270–286 CrossRef CAS PubMed.
- Q.-Q. Xu, C.-J. Zhang, X.-Z. Zhang, J.-Z. Yin and Y. Liu, J. Supercrit. Fluids, 2012, 62, 184–189 CrossRef CAS PubMed.
- B. Cornelio, G. A. Rance, M. Laronze-Cochard, A. Fontana, J. Sapi and A. N. Khlobystov, J. Mater. Chem. A, 2013, 1, 8737–8744 CAS.
- A. Primo, A. Corma and H. García, Phys. Chem. Chem. Phys., 2011, 13, 886 RSC.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 CAS.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- Z. Zhang, L. Li and J. C. Yang, J. Phys. Chem. C, 2013, 117, 21407–21412 CAS.
- C. T. Campbell and J. R. V. Sellers, Faraday Discuss., 2013, 162, 9–30 RSC.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- J. H. Fendler, Nanoparticles and Nanostructured Films: Preparation, Characterization, and Applications, John Wiley & Sons, 2008 Search PubMed.
- J. A. Schwarz, C. I. Contescu and K. Putyera, Dekker Encyclopedia of Nanoscience and Nanotechnology, CRC Press, 2004 Search PubMed.
- D. A. Walker, B. Kowalczyk, M. O. de la Cruz and B. A. Grzybowski, Nanoscale, 2011, 3, 1316–1344 RSC.
- D. J. Bray, S. G. Gilmour, F. J. Guild, T. H. Hsieh, K. Masania and A. C. Taylor, J. Mater. Sci., 2011, 46, 6437–6452 CrossRef CAS.
- J. T. Miller, M. Schreier, A. J. Kropf and J. R. Regalbuto, J. Catal., 2004, 225, 203–212 CrossRef CAS PubMed.
- M. Lashdaf, T. Hatanpää, A. O. I. Krause, J. Lahtinen, M. Lindblad and M. Tiitta, Appl. Catal., A, 2003, 241, 51–63 CrossRef CAS.
- M. L. Toebes, J. A. van Dillen and K. P. de Jong, J. Mol. Catal. A: Chem., 2001, 173, 75–98 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10736e |
| This journal is © The Royal Society of Chemistry 2014 |