
A highly sensitive method for simultaneous determination of the quaternary ammonium pesticides chlormequat and mepiquat in pears and potatoes by modified QuEChERS-high performance liquid chromatography-tandem mass spectrometry†
Abstract
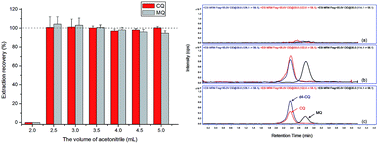
A highly sensitive high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method for simultaneous determination of the quaternary ammonium pesticides chlormequat (CQ) and mepiquat (MQ) in pears and potatoes was developed and fully validated. The modified QuEChERS method was employed for sample preparation. A hydrophilic interaction liquid chromatography (HILIC) column was used for the chromatographic separation of highly polar analytes. The detection was performed by a triple quadrupole mass spectrometer with an electrospray ionization (ESI) source in positive ion mode by multiple reaction monitoring (MRM). The detailed fragmentation mechanisms of targeted analytes in MS/MS system were studied on the theoretical level using density functional theory (DFT) calculations. The limits of detection (LODs) were 0.021 μg kg−1 and 0.21 μg kg−1 for CQ and MQ, respectively. The mean recoveries of CQ and MQ were in the range of 83.4–119.4% with RSD less than 7.0%. The developed method was applied to the analysis of CQ and MQ in actual samples from different retail outlets in China, implying its potential in fast monitoring of CQ and MQ residues.
 Please wait while we load your content...
Please wait while we load your content...

