
Simultaneously enhancing strength and toughness for impact polypropylene copolymers by regulating the dispersed phase with high density polyethylene
Biwei Qiu,
Feng Chen,
Yonggang Shangguan*,
Lina Zhang,
Yu Lin and
Qiang Zheng*
and
Qiang Zheng*
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, People’s Republic of China. E-mail: shangguan@zju.edu.cn; zhengqiang@zju.edu.cn; Fax: +86 571 8795 2522; Tel: +86 571 8795 2522
First published on 24th October 2014
Abstract
By introducing high density polyethylene (HDPE) into the dispersed phase of impact polypropylene copolymers (IPCs), the morphologies of IPC/HDPE blends were regularly tailored and consequently the tensile and impact properties were simultaneously improved. Morphological observations showed a series of multilayered core–shell dispersed particles when the content of HDPE was less than 40%, while the continuous network structure was observed beyond 40%. With an increase in the content of HDPE, the size of the core increased and the number of dispersed particles with incomplete encapsulated polyethylene (PE) cores rose. More valid ‘bridges’ made up of segmented ethylene–propylene copolymer (sEbP) appeared and connected the PE core and polypropylene (PP) matrix. Meanwhile, co-crystallization occurred in the core phase, between long ethylene chain segments of the joined HDPE and sEbP in multi-component IPCs. The increased HDPE in blends reduced defective co-crystals, and in turn led to a thicker average lamellar thickness and thinner amorphous thickness of PE. Partial inserted ethylene–propylene random sequences are constrained by narrowed PE amorphous layers. Hence, the connection between the PP matrix and the dispersed phase was strengthened by co-crystals, ‘bridges’ and restriction effects. The tensile strength of the blends was slightly enhanced with an increase in HDPE, while the greatly improved toughness was achieved at a HDPE content of 30 wt% and kept constant with more HDPE. Thus, the interactions rather than core–shell phase morphology are regarded as the predominate factor for the excellent properties.
1. Introduction
In recent decades, impact polypropylene copolymers (IPCs)1,2 synthesized by a two-step polymerization including bulk polymerization of propylene and then gas-phase copolymerization of ethylene and propylene, have been widely applied in many industrial fields due to the improved toughness compared with ordinary isotactic propylene (iPP). IPCs have been experimentally characterized to be a multiphase polymer system which consist of three components: homopolymer polypropylene (HPP) as the matrix, an amorphous ethylene propylene random copolymer (EPR) and a segmented ethylene–propylene copolymer (sEbP) forming the dispersed phase.2–4 Here, the chains of the sEbP component contain not only a crystallizable polyethylene segment (sEbP-E) and a polypropylene segment (sEbP-P), but also some ethylene–propylene random sequences.2,5 It is generally accepted that the toughness enhancement for this multicomponent product is attributed to the special dispersed phase with a multilayered core–shell structure, where the inner core is mainly composed of sEbP-E, the intermediate layer is EPR and outer layer is sEbP-P.2,6 Compared with ordinary blends with toughened PP by soft rubber particles, improved interfacial adhesion between the dispersed particles and the matrix in IPCs guarantees better energy transmission and dissipation. However, the introduction of various copolymers in IPCs brings about the undesired loss of stiffness and strength due to a wider distribution of molecular weight, as most of the reported toughened PP is modified with rubbers.7–9 Thus, how to fabricate a polymeric product with simultaneously enhanced strength and toughness is still a challenge of IPC development.To obtain polymeric materials with a stiffness balance or both excellent properties, increasing the rigidity of the dispersed particles (add inorganic/organic rigid particles or a crystalline polymer) is regarded as an efficient method.10,11 Many factors are related to the final mechanical properties of these modified polymers, such as the size of the dispersed particle, interfacial adhesion, crystallization behavior and so on.12 Usually, some inorganic fillers, such as calcium carbonate (CaCO3) and silica (SiO2), are used to improve the stiffness and impact toughness of a polymer,13–19 since they can act as points of stress concentration and induce massive crazing to enhance energy dissipating processing. Some of the nanoscale particles even play the role of nucleation agent and influence toughness through reducing spherulite size.20 However, due to weak interfacial adhesion between the dispersed particles and the matrix as well as bad dispersion, it is still difficult to achieve good toughness.20,21 In addition, incorporation of another crystalline polymer in the modified polymer can also improve toughness without serious sacrifice of stiffness and various toughening mechanisms have been presented.10,22–25 It was reported that the introduction of polyethylene into PP brought dramatic improvement in toughness23,24 and J. C. Feng et al.23 believed that it should be mainly ascribed to the reduced spherulite size of PP by the introduction of HDPE. B. Na et al.10 fabricated nylon-6/poly(vinylidene fluoride) (PVDF) blends and found that plastic deformation of the matrix evoked by PVDF particles was responsible for the toughness enhancement. Kurauchi et al. proposed a concept of cold drawing that the plastic deformation of brittle particles themselves is the major reason of toughening, rather than shear yielding of the matrix induced by dispersed particles.25 However, the poor interfacial adhesion between dispersed particles and the matrix in modified polymers mentioned above is an unavoidable problem. To improve the interfacial adhesion, compatible components were generally introduced into the above blends and consequently better toughness improvement was obtained.22,26,27 Morphology observation showed that special dispersed particles with a core–shell structure formed in such blends and they were thought to be the major contributor of improvement in toughness.
As reported previously, the dispersed phase with a multilayered core–shell structure in IPCs can be rebuilt by blending components of HPP/EPR/EbP, and the morphology and thicknesses of shells can be regulated through varying the composition ratio.28 Far more important was that these blends present different dynamic mechanical behavior. Considering the fact that the HDPE component can enter into the core of the dispersed particles of the IPC when blending IPC with minor HDPE,29 introducing HDPE into IPCs may be a practical and effective approach to regularly control the complex microstructure of a IPC and in turn improve its mechanical properties, as a result the new IPC with greater excellent strength and toughness may be achieved. Thus, in this work, we systemically investigated the effect of the introduction of HDPE on phase morphology evolution and the interaction of components, as well as the toughening mechanism in IPC/HDPE blends.
2. Experimental
2.1 Materials and sample preparation
IPC (SP179, Mw = 1.74 × 105, Mw/Mn = 3.96) was supplied by Sinopec Qilu Petrochemical Co. of China and HDPE (5000 s, Mw = 2.83 × 105, Mw/Mn = 5.87) was purchased from Sinopec Beijing Yanshan Company. IPC and HDPE were melt mixed on a torque rheometer (XSS-300, Kechuang Co. Ltd., China) at 180 °C for 10 min and with a roll speed of 60 rpm. The antioxidant (0.2 wt%), Irganox 1076, was introduced into blends to avoid oxidative degradation during mixing. IPC/HDPE blends with compositions of 100/0, 95/5, 90/10, 80/20, 70/30, 60/40, 50/50 and 0/100 were prepared respectively. To simplify their name, they are successively labeled as E0, E5, E10, E20, E30, E40, E50 and E100 respectively. The number behind E stands for the content of HDPE in the blend. After that, all blend samples were compression molded at 180 °C for 8 min. The standardized dumbbell-shaped samples with a thickness of 1.75 mm, width of 4.5 mm and length of 25 mm were prepared and tensile tests (23 °C) were carried out according to GB/T 1040 with a speed of 25 mm min−1. Notched charpy impact strength was measured on a notch impact instrument (MTS systems Co. Ltd., China) according to GB/T 1043.1-2008. The size of the rectangular specimens was 80 × 10 × 4 mm3 and V-shaped notches of about 45° were cut (2 mm depth) by using a semi-automatic notcher before measurement.2.2 Scanning electron microscopy (SEM)
In order to elucidate the complex phase structure, partial samples were adequately etched in xylene to remove the EPR and EbP phases at 50 °C for 4 h and at 100 °C for 0.5 h respectively.2 After drying in an oven at 60 °C for 12 h, all cryogenically fractured surfaces of the blend samples were sputter-coated with gold powder before being observed by SEM (Hitachi, S-4800).2.3 Differential scanning calorimetry (DSC)
A DSC (Q100, TA Instruments Corporation) with nitrogen as the purge gas was used to probe the crystallization and melting behavior of the blend samples. Each sample, about 8.0 mg, was sealed in an aluminum pan to heat to 210 °C at a rate of 3 °C min−1 and the corresponding endothermic curve was recorded as its melting trace.2.4 Wide-angle X-ray diffraction and small-angle X-ray scattering (WAXD/SAXS)
WAXD patterns of the film samples were obtained on a D/Max-2550 PC X-ray diffractometer with the Cu Kα radiation at room temperature. The wavelength (λ) of the X-ray beam was 1.54059 Å. In this study, the continuous scanning angle (2θ) ranged from 3° to 40° at a voltage of 40 kV and a current of 250 mA. The d-spacing of the sample was scanned at 5° min−1, with a step of 0.02°. The overall crystallinity (Xc) was calculated according to the following equation:30,31
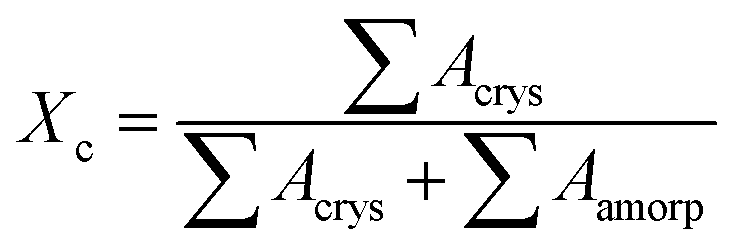 | (1) |
After the amorphous background has been extracted, Acrys and Aamorp are the areas of the fitted crystal peaks and amorphous contribution peak respectively.
SAXS experiments were performed on a XEUSS SAXS/WAXS SYSTEM (French, XENOCS SA) with Cu Kα radiation and the wavelength of the X-ray radiation was 1.5411 Å. The generator was operated at 30 kV and 650 mA. The thickness for all specimen sheets was around 0.75 mm. The distance between the sample and detector was 2522 mm. The length of the scattering vector q in the SAXS measurements ranged from 0.05 to 1.24 nm−1. The SAXS intensity was collected with a two-dimensional gas-filled wire detector and each SAXS pattern was collected within 600 s. The measured SAXS intensity was calibrated for background scattering by direct model fitting of the data in Matlab, and then the one-dimensional correlation function K(z) was calculated by cosine Fourier transformation, seen in eqn (2). The long period L was calculated from the peak position in the curve of the one-dimensional correlation function.
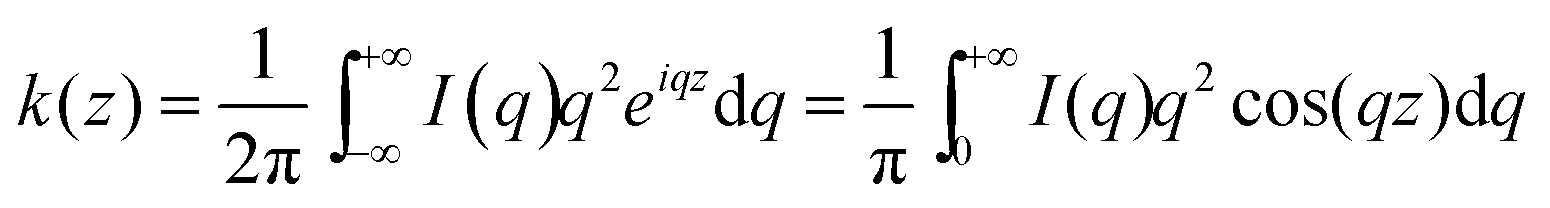 | (2) |
2.5 Dynamic mechanical analysis (DMA)
The dynamic mechanical behavior of the blend samples was investigated in single cantilever mode on a Q800 DMA (TA Instruments Corporation, USA). The dimensions of the samples were about 17.5 mm × 12.7 mm × 1.5 mm. Dynamic loss (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) was determined at a frequency of 1 Hz and a heating rate of 3 °C min−1, as a function of temperature (from −130 to 140 °C).
δ) was determined at a frequency of 1 Hz and a heating rate of 3 °C min−1, as a function of temperature (from −130 to 140 °C).
3. Results and discussion
3.1 Phase morphology
SEM images of the etched fracture surface for IPC and IPC/HDPE blends are given in Fig. 1. It is seen that the morphologies of the blend samples can be divided into two categories after the introduction of HDPE into IPC. When the HDPE content is no more than 40% in weight, the dispersed phase of the blend samples is in a particle shape; on the contrary, when the HDPE content is greater than 40%, a continuous dispersed phase appears. Here, the holes and hollowed parts represent removed EPR domains as only this component can be completely dissolved in xylene at 50 °C.2 Note that the dispersed particles in IPC/HDPE blends have a similar core–shell structure as pure IPC and only the size of core enlarges with the increase of HDPE. These morphological results indicate that most of the HDPE component enters into the core domain. Fig. 1(a) gives the morphological structure of the IPC, and two kinds of holes with and without inclusions are observed, in accordance with previous results.29 The former comes from dispersed particles whose PE cores are encapsulated incompletely by a rubber shell, so the core phase can’t be brought out with an etched EPR shell. As for the latter, it may result from two cases: the PE core is completely encapsulated by EPR or pure EPR particles. As seen in Fig. 1, since the size of the core increases monotonically with increasing the content of HDPE in the blends, the concentration of EPR declines and consequently the EPR cannot encapsulate bigger PE cores entirely. As a result, solely holes reduce and holes with inclusions increase significantly. As the content of HDPE increases to 40 wt%, phase morphology of the continuous dispersed phase begins to appear, resulting from the integration of an increasing dispersed domain. In this case, the EPR component is still located at the interface between HDPE and HPP. | ||
| Fig. 1 SEM micrographs of the etched fracture surface for IPC and IPC/HDPE blends after removing the EPR component. (a) E0; (b) E5; (c) E10; (d) E20; (e) E30; (f) E40; (g) E50; (h) E100. | ||
To understand the morphological features of the blend samples, the samples were subsequently etched in xylene at 100 °C to remove all ethylene–propylene copolymers (sEbP and EPR components) adequately. Fig. 2 gives SEM images of some samples with typical morphological features including neat IPC, the blends containing dispersed particles and a continuous dispersed domain. The remains are crystallizable HPP and PE components, which cannot be dissolved in these conditions.2,5 Compared with Fig. 1, all new holes and broader gaps in Fig. 2 are ascribed to the etched sEbP component. For pure IPC, the bigger holes appear but their inclusions disappear (Fig. 2(a)), further suggesting that the sEbP component locates at both the outer layer and core phase of the core–shell dispersed particles. As for the IPC/HDPE blends, the increasing gap is also attributed to the removal of the sEbP-P located between the EPR shell and the matrix. Fig. 2(b) clearly demonstrates that the dispersed phase in IPC/HDPE blends consists of similar multilayered core–shell particles as in pure IPC when the HDPE content is less than 40%. For sample E50, the enlarged boundary gap in blends with a bicontinuous morphology (Fig. 2(c)) is also stemmed from sEbP, which can improve interfacial interaction.
 | ||
| Fig. 2 SEM micrographs of the etched fracture surface of IPC and its blends after removing all EPR and EbP components. | ||
The above results show that when a small quantity of HDPE as a secondary component is blended with IPC, the blend samples also present the multilayered core–shell dispersed particles and the size of the core can be regulated by HDPE content. Generally, interfacial tension of the components is presumed to be the main driving force of morphology evolution for a multi-component system.32–34 Since HPP and HDPE are completely incompatible, one can add compatibilizer (such as block polymers by copolymerization) into them to reduce interfacial tension. The compatibilizer is usually distributed around the interface due to its good compatibility with both components.32,33,35–37 Therefore, the shell which is comprised of compatibilizer can encapsulate the minor component to form core–shell dispersed particles. Based on Palierne’s emulsion model and Gramespacher–Meissner analysis, K. Cornelia et al.33 verified that the core–shell morphology in high impact propylene–ethylene copolymers (HIPP), with PE lamellae situated totally inside EPR inclusions, was determined by viscosity and compatibility of the components. M. Hemmati et al.36 also found that the experimental result of core–shell type morphology for HDPE encapsulated by rubber was in good agreement with the theoretical predictions for PP/rubber/PE ternary blends. Thus, the multilayered core–shell morphology in the IPC/HDPE blend is in a thermodynamically stable state under certain conditions.
3.2 Crystallization and interaction of components
After introducing HDPE into the IPC, the influence on the crystallization and interaction of multi-components in IPC/HDPE blends were investigated.To detect crystalline information of the blend samples, their WAXD patterns were studied (Fig. 3). Diffraction peaks (2θ) at angles of 14.0°, 16°, 16.8°, 18.6°, 21.2° and 21.9°, corresponding to the (110), (300), (040), (130), (111) and (131) & (041) lattice planes, belong to crystals of HPP. The diffraction peak for the (300) lattice plane results from β crystals and others from α-crystals. Besides, (110) and (200) lattice planes at 21.6° and 24° are two preferential directions for the crystal growth of PE.38,39 With the increase in HDPE content and the decrease in IPC simultaneously, most characteristic peaks of HPP, such as at (110), (300), (130) and (131) & (041) lattice planes, first increase and then decrease, while the diffraction intensity of both the lattice planes of PE strengthens. These changes are strongly dependent on the contents of crystalline polymers. The only exception is that, PP crystals along the (040) lattice plane exhibit a slow increase with the increase of PE. The total crystallinity (Xc) for PP and PE were calculated according to the method mentioned in the experimental section and the results are listed in Table 1. The total crystallinity of PP (Xc,PP) ascends in the beginning and then descends with the decrease of IPC content, rather than in proportion. This result is in accordance with the changes of most diffraction peaks of PP. The increment of Xc,PP may result from the introduction of HDPE. J. C. Feng et al.23 once reported the decreased spherulite size of PP and the increased concentration of PP intercrystalline due to blending HDPE with PP. In other words, HDPE can promote crystallization of PP to some extent.
 | ||
| Fig. 3 WAXD patterns of IPC and IPC/HDPE blends. | ||
| Samples | E0 | E5 | E10 | E20 | E30 | E50 | E100 |
|---|---|---|---|---|---|---|---|
| Xc,PP | 0.229 | 0.245 | 0.254 | 0.231 | 0.199 | 0.161 | 0 |
| Xc,PE | 0.065 | 0.087 | 0.115 | 0.134 | 0.141 | 0.209 | 0.476 |
In this work, although most of HDPE component lies in the core domain, some PE cores cannot be encapsulated entirely by the ethylene–propylene copolymer when HDPE content gradually increases. Compared with pure IPC, more ‘bridges’ connecting the core domain with the PP matrix can be observed, as indicated by the arrows in Fig. 4. Since the EPR component has been removed in these samples, considering the poor compatibility between HDPE and PP, the ‘bridge’ can only be comprised of sEbP which has a better compatibility with either PE or HPP, as documented by previous reports.2,28 It is noticed that in pure IPC only a spot of sEbP acts as the compatibilizer to form visible ‘bridges’ while most of the sEbP consists of the core domain. With the introduction of HDPE into the core domain, some sEbP in the core are pushed outwards to the interface between the core and the HPP matrix to form ‘bridges’, although the total content of sEbP should descend with the decrease of IPC content in blends. These results may result in a stronger link between the dispersed particles with the HPP matrix.
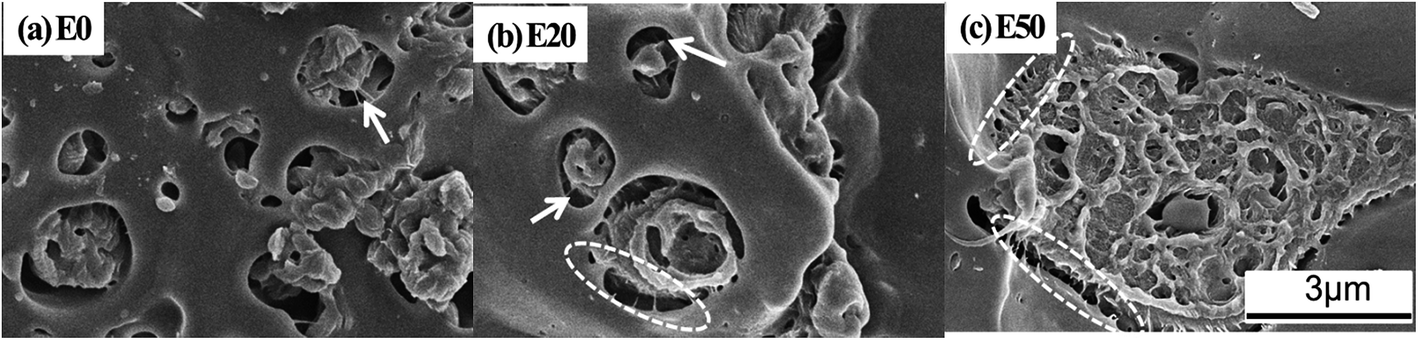 | ||
| Fig. 4 SEM micrographs of the etched fracture surface of IPC and its blends after removing the EPR component. | ||
DSC melting curves of blend samples are given in Fig. 5. The double endothermic peaks at about 160 °C and higher temperatures correspond to the melting of PP crystals while the peaks at about 130 °C belong to PE crystals. It is noticed that although the WAXD results of the blend samples confirm the existence of β-PP crystals, the peak temperatures of the PP crystals on DSC traces are no less than 160 °C, which are much higher than the melting temperature of β-PP crystals (141–153 °C).40 Considering the fact that β-PP is metastable and is apt to transform into the stable α form when being heated,41 it is reasonable that the double melting peaks of PP (labeled as peak PP1 and PP2) should be attributed to defective crystals and perfect crystals of α-PP respectively. It can be inferred that the prominent PP2 peaks at a higher temperature reflect most PP crystals in the matrix. As for the weak PP1 peaks, they may mainly result from imperfect α-crystals at the interfacial domain affected by the introduction of HDPE. Compared with neat IPC, the peak temperatures for PP1 in all IPC/HDPE blends are higher, which may be due to the promoting effect of HDPE on the crystallization of PP to some extent. In addition, the peak temperature of PE rises with increasing HDPE content in blends. Two components in the blends, HDPE and sEbP-E, offer long chain segments of ethylene. It has been demonstrated in previous investigations2,29 that the melting peaks would stay at characteristic locations respectively for ‘unmixed samples’, and their crystallizable components would not influence each other. Here, the dependence of melting peak temperature for PE on composition suggests that certain interactions exist between the HDPE and sEbP-E component. In dispersed particles, both HDPE and sEbP-E locate at the core domain. Similar ethylene segments can rearrange regularly together to form PE co-crystals. The same result is in good accordance with the previous work.29 The higher the melting temperature is, the more perfect the PE crystals are. Fig. 6 gives the morphology of the PE domain for pure HDPE E100 and blend samples of E20 and E50 after removing most amorphous components (EPR and sEbP). The layered-structure PE domain consisted of PE lamellae can be observed in all of them. Under the same magnification, denser and better-organized layers are found in the samples with a higher content of HDPE, especially in pure HDPE.
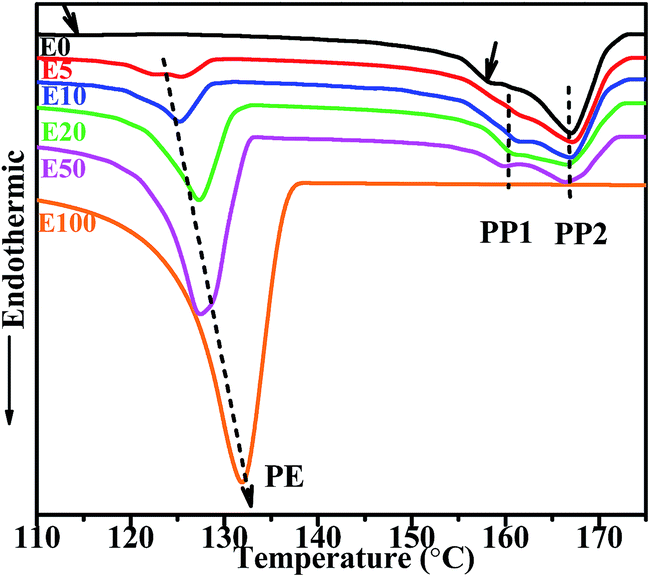 | ||
| Fig. 5 DSC endotherms of IPC and its blends at a heating rate of 3 °C min−1. | ||
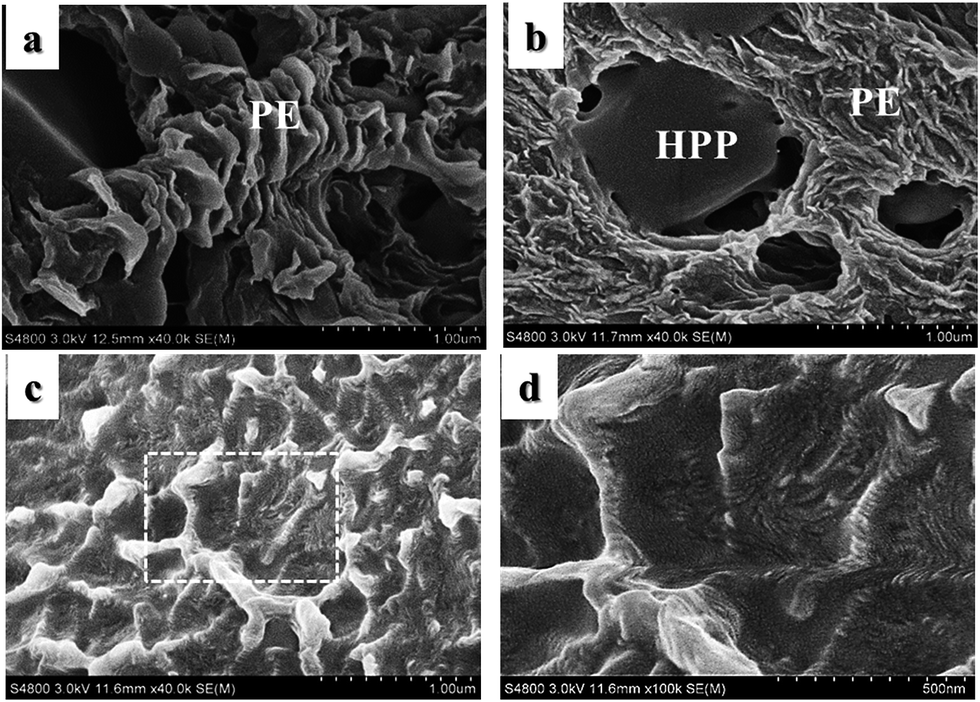 | ||
| Fig. 6 SEM micrographs of the PE domain in IPC/HDPE blends and pure HDPE after etching in xylene at 100 °C for 0.5 h. (a) E20, (b) E50, (c) E100 for pure HDPE and (d) is a partially enlarged image of the dashed box in (c). | ||
To further explore the interaction of the components, SAXS measurement for the IPC/HDPE samples was conducted. Firstly, two-dimensional SAXS patterns over a pie-shaped area were integrated to obtain the one-dimensional SAXS intensity profile I, which is a function of the scattering vector q.42 Lorentz correction (I × q2) is applied to deal with scattering intensity as the crystals consist of layer-like lamellae with a spherical symmetrical structure. Then one-dimensional electron density correlation function k(z) can be derived from the corrected scattering intensity I(q) through eqn (2). Fig. 7 illustrates the Lorentz corrected intensity profile and the SAXS correlation functions for the film samples. From Fig. 7(a), one peak located at about q = 0.25 nm−1 is obtained for HDPE while that at about q = 0.5 nm−1 is found for neat IPC, attributed by PE and PP lamellae separately. Meanwhile, double peaks close to them respectively are observed in IPC/HDPE blends, indicating the coexistence of two crystals. The long period (L) can be evaluated by L = 2π/qmax, where qmax is the position of the maximum intensity of the corrected scattering curve. Besides, L can be obtained by the z value of the first maxima of k(z) too. The values of L estimated from both methods are very close. In a non-ideal two-phase system, L is assumed to be the sum of average thicknesses of lamellae (Lc), amorphous layers (La) and transition zones (Ltr) (L = Lc + La + 2Ltr).43,44 Compared with the relaxed mobile amorphous fraction (MAF), the transition zone is termed to the ordered rigid amorphous fraction (RAF), distributed at the interface between the crystal and mobile amorphous layers. To simplify the analysis, the layers of MAF and RAF could be considered as a whole, that is LA = La + 2Ltr. Usually, the values of structural parameters (Lc and Ltr) can be determined by the curve of k(z) (as indicated by arrows in Fig. 7(b)) and then La equals the subtraction of Lc and 2Ltr from L.45 However, the mixing crystals of PP and PE would lead a large uncertainty and error in the calculation of Lc and Ltr by this method. It is noted that Lc could also be estimated by melting temperature investigated through DSC.46,47 Though there are differences in Lc measured by SAXS and DSC, the variation trends are consistent with each other.48 So in this work, the Lc is just analyzed qualitatively by DSC and the thickness of the total amorphous layer (LA) is obtained by the subtraction of Lc from L. The values of L for different samples are listed in Table 2.
 | ||
| Fig. 7 Lorentz corrected SAXS intensity profiles (a) and curves of the one-dimensional correlation function (b) for IPC and its blends. | ||
| Samples | E0 | E20 | E30 | E50 | E100 |
|---|---|---|---|---|---|
| LPP (nm) | 12.75 | 12.61 | 12.54 | 12.4 | — |
| LPE (nm) | — | 23.56 | 23.15 | 22.68 | 21.65 |
The values of the long period LPP and LPE for neat IPC and HDPE are about 12.75 and 21.65 nm, respectively. With increasing HDPE content in IPC/HDPE blends, the LPP decrease slightly, whereas the decrease of LPE is distinct. Based on the relationship between lamellar thickness and melting temperature,46,47,49 the average Lc values of PP lamellae (Lc,PP) almost keep constant as the respective melting points of PP are identical in the blend samples (Fig. 5). But the Lc values of PE lamellae (Lc,PE) thicken monotonously with increasing HDPE content. As a result, LA,PP and LA,PE both tend to decrease (LA = L − Lc), indicating a reduced gap between lamellar crystals: denser PE lamellae have organized. In this case, ethylene segments with a high concentration can fold regularly with little infiltration by relaxed ethylene segments and ethylene–propylene random segments. Generally speaking, it is difficult for other chains to insert into PE lamellae. It should be pointed out that chains of the sEbP component in IPCs have been confirmed to contain three sequences: a long ethylene segment, a long propylene segment and an ethylene–propylene random sequence.5 When co-crystallization occurs between the ethylene-rich sEbP and HDPE component, the ethylene–propylene random sequences in the sEbP chains are brought into adjacent amorphous zones. Consequently, the interaction between copolymers and HDPE has been enhanced.
Simultaneously, the change in amorphous thickness can be characterized with DMA measurement. The temperature-dependent loss factor (tanδ) is illustrated in Fig. 8. Three loss peaks are obtained in IPC and IPC/HDPE blends, belonging to glass transitions of the PE, EPR and PP components from a low to high temperature, respectively.2 The PE transition peak located at about −100 °C is found for pure HDPE. The intensity of the PE tanδ peak increases with more HDPE, indicating enhancement of the mobile amorphous fraction in the quantity. Similarly, the intensity of the PP tanδ peak is almost proportional to IPC content in blends too. It is noted that the glass-transition temperatures (Tg) of PP and PE are maintained though the LA,PP and LA,PE mentioned above tend to reduce. It can be explained that composition directly affects the amount of amorphous PP component and PE domain in the core but has little effect on molecular mobility and their free volume. As for the loss peak of EPR, the peak area tends to decrease with increasing HDPE content, which should be attributed to the decreased proportion of amorphous EPR component in these blends. But it is interesting that the transition peak of EPR shifts to a higher temperature, indicating the weakened mobility and shrinking free volume. However, considering the fact discussed above that the decreased LA,PE results from thickened lamellae and less infiltration of ethylene–propylene random sequences, the shift of the EPR transition peak may be explained reasonably. That is to say, ethylene–propylene random chain segments are constrained further by narrowed gaps between lamellae and in turn it makes a rise in the EPR transition peak. This result is further evidence of the enhancement of connection between the ethylene–propylene copolymer shell and PE core phase.
 | ||
| Fig. 8 Temperature dependence of the mechanical loss factor (tanδ) of IPC/HDPE blends at a frequency of 1 Hz and a heating rate of 3 °C min−1. | ||
3.3 Mechanical properties
As discussed above, different morphologies of blend samples can be obtained, depending on the amount of introduced HDPE. When HDPE content is no more than 40%, most of the HDPE is encapsulated by a copolymer shell to form the core domain and only the sizes of dispersed particles are regularly changed. This is understandable that the different microstructures will finally influence subsequent mechanical properties, so the mechanical properties are investigated by the tensile test and notched charpy impact test and discussed in detail below.The engineering stress–strain curves of IPC/HDPE blends are shown in Fig. 9(a). Fracture energy and toughness can be expressed by the area under the curve in some extent. Both yield strength (δy) and elongation at break (εb) of the blend samples raise slightly, compared with neat IPC. However, for blends E40 and E50 with a bicontinuous phase morphology, both δy and εb sharply increase and they almost approach that of neat HDPE. These results show that the stiffness of the blend has been strengthened by the introduction of HDPE, rather than sacrificing the stiffness and strength like most toughening systems.7–9 In this case, the strength is determined by two factors: (i) adhesion between the matrix and dispersed particles; (ii) deformation ratio of the matrix and dispersed phase under the same tension. Factor (i) is dependent on the interaction between components. As discussed above, the joint effect on the HPP matrix and dispersed particles is strengthened with increasing content of HDPE due to the increase in sEbP bridges and co-crystals, as well as an enhanced interaction of the PE core and the intermediate EPR layer. In addition, factor (ii) has a strong influence on deformation and yield. Generally, crystalline polymers (such as the PP matrix and PE core here) are able to accommodate large elongation under a tensile stress, which is attributed to cold drawing.25 They deform in a ductile manner and absorb a significant amount of energy. As in dispersed particles composed of elastomeric materials or amorphous copolymers, large deformations can also occur, making them suitable for employment in the dissipation of energy. But weak adhesion between the HPP matrix and dispersed phase is not beneficial to deform simultaneously, and defects and voids would appear at their interface during stretching. After interfacial reinforcement by incorporation of HDPE, the stress can be effectively transferred to the dispersed phase. Meanwhile, the crystallized PE core generates shear deformation accompanied by massive energy dissipation.
 | ||
| Fig. 9 (a) Typical engineering stress–strain curves and (b) notched charpy impact strength for IPC and its blends. | ||
To verify our deduction, SEM images of blends before and after the tensile test at the same magnification are shown in Fig. 10. The fracture surface is chosen at the middle section which is parallel to the tensile direction (directed by the red arrow in Fig.10(a)). The elongated holes with a few inclusions have been observed in IPC, originated from the nonuniform stress transmission between the matrix and dispersed phase. The soft core–shell particles peeled off the matrix because of weak adhesion. With the introduction of HDPE and enhanced connections at the interface, elongated dispersed particles including stretching of the PE core (marked by a dashed box and arrow in Fig. 10(b)–(d)) appear, because the crystalline PE core can deform with the HPP matrix simultaneously through cold drawing and shearing deformation. Meanwhile, the vaguer boundary indicates an enhanced interfacial effect and a more slender dispersed phase signifies greater strain at a higher HDPE content. Apparently, enhanced interaction and shearing deformation of the PE domain in the core both favor energy dissipation. In particularly, the extremely high strength in blends with a bicontinuous morphology may be due to a interconnected HDPE phase which can be stressed homogeneously, similarly in neat HDPE.
 | ||
| Fig. 10 SEM micrographs of the fracture surface before (small inserted images, EPR was etched) and after tensile test (corresponding large ones, unetched). | ||
The fracture resistance of the blends has been further detected by the notched charpy impact measurement at room temperature and the results are shown in Fig. 9(b). Neat IPC and HDPE both exhibit poor impact strength, which is lower than 20 kJ m−2. Surprisingly, the impact strength starts to increase dramatically after the introduction of 10 wt% HDPE. A maximum impact strength is obtained at 30 wt% HDPE content in blends and then is kept constant when bicontinuous morphology forms. The increment which is more than three times in impact strength has been achieved.
Morphological observation of impact-fractured surface is also favorable to understand the toughening mechanism, so fracture surfaces of IPC/HDPE blends were demonstrated in Fig. 11. When the HDPE content is lower than 10 wt% (Fig. 11(a) and (b)), a smooth matrix surface and cavitation (inserted images) can be observed on the whole fractured surface, indicating the typical morphology of a brittle-fracture mode. A few drawing dispersed particles with stretchy cores occur, enhancing the impact energy absorption by deformation. As the HDPE content reaches 10 wt% (Fig. 11(c)), the deformed dispersed phase along the impact direction at a small angle increases. Generally, cavitation happens in soft rubber particles, which act as stress concentrators to generate crazes or even cause shear yielding of the crystalline phase. By contrast, the occurring of shear yielding or plastic deformation of the crystalline phase is more efficient for the consumption of energy. Here, the deformed dispersed particles with PE cores are exactly contributed by shear yielding of PE crystals. In the rest of the cases, blends with more HDPE (Fig. 11(d)–(f)), large plastic deformation is initiated to consume the energy effectively and excellent impact strengths are acquired. Especially in bicontinuous morphology blends, stress transfer is also continuous which results in a maximum on impact strength. It is noted that the big change in strength starts from E10 but the large increase in strain starts from E30. It may result from different phase morphologies and interfacial interaction. When the HDPE content in the blend increases to 10 wt%, the size of the cores and whole dispersed phases with a core–shell structure start to increase distinctly. Besides, the interaction between the HPP matrix and dispersed particles also starts to strengthen by co-crystal and sEbP ‘bridges’. As a result, the strength increases because fracture energy can be effectively transferred and dissipated by a reinforced connection and the deformed dispersed phase. However, at this moment the dispersed phases are still easy to be peeled off from the matrix because of a different deformation ratio. Until the HDPE content in the blend reaches 30 wt%, a large number of dispersed phases with a core–shell structure get close to each other and approach a continuous structure. Then the matrix and nearly interconnected dispersed phase can be stressed homogeneously and a large strain is achieved through cold drawing and shearing deformation simultaneously.
 | ||
| Fig. 11 SEM micrographs of the etched fracture surface after impact test for IPC/HDPE blends. The inserted ones are corresponding unetched images. | ||
Considering that either the blend samples with core–shell dispersed particles or the blend samples with a bicontinuous morphology present good enhancements on tensile property and impact strength, a conclusion may be drawn that the core–shell structure of the dispersed particle in IPC/HDPE blends may not be the predominant factor in simultaneously enhancing strength and toughness. Actually, the interaction among components strengthens the connection between the matrix and dispersed particles so that fracture energy can be transferred and dissipated effectively. Many works15,27,50 show that toughness has been greatly improved by core–shell phase morphology, due to the reinforced connection between the core and matrix by the shell. In these blends, the copolymers in IPC play important roles in enhancing interactions and a good toughness–stiffness balance is acquired compared with common blends of PP/HDPE.23
4. Conclusions
Through melt blending IPC and HDPE, the morphology of the multilayered core–shell dispersed phase and the size of the inner PE core are tailored. The stiffness and size of the core phase increase with the introduction of more crystalline HDPE, expecting to get a toughness–stiffness balance or both excellent properties. A schematic diagram in Fig. 12 shows the morphology evolution of dispersed phases after the introduction of HDPE and the corresponding deformation mechanism. With an increase of HDPE content in blends, more and more large cores can’t be encapsulated completely. Some sEbP in the core is pushed outwards to the interface between the core and the HPP matrix to form ‘bridges’. Here, the core phase is composed of sEbP-E and HDPE. Similar ethylene chain segments in them form co-crystals and the thickening PE lamellae restrict imported ethylene–propylene copolymer further. Both of them are beneficial to reinforce the interaction between the core phase and intermediate EPR layer. | ||
| Fig. 12 Schematic diagrams representing the effects of HDPE content on morphology and the deformation mechanism of IPC/HDPE blends. | ||
The mechanical property shows that the toughness is effectively enhanced by the introduction of HDPE into IPC. The reinforced connection between the dispersed phase and matrix rather than core–shell morphology is the basic reason for the great improvement of the properties. Accompanied by increasing interaction among the matrix, intermediate EPR layer and PE core, the stress can be transferred effectively and induce shearing deformation of the PE core, consuming the energy significantly.
The work provides us not only with a deep insight for understanding the toughening mechanism for multiphase systems, but also a new thought to toughen IPC without sacrificing rigidity by introduction of crystalline polymers. The applied range of IPC as a high-performance material with balanced mechanical properties is greatly broadened.
Acknowledgements
This work was supported by National Nature Science Foundation of China (no. 51173157, 51173165), the Fundamental Research Funds for the Central Universities (no. 2013QNA4048) and Nature Science Foundation of Zhejiang Province (no. Y4100314).References
- S. Cheruthazhekatt and H. Pasch, Macromol. Symp., 2014, 337, 51–57 CrossRef CAS.
- C. H. Zhang, Y. G. Shangguan, R. F. Chen, Y. Z. Wu, F. Chen, Q. Zheng and G. H. Hu, Polymer, 2010, 51, 4969–4977 CrossRef CAS PubMed.
- S. Cheruthazhekatt, T. F. J. Pijpers, G. W. Harding, V. B. F. Mathot and H. Pasch, Macromolecules, 2012, 45, 5866–5880 CrossRef CAS.
- S. Cheruthazhekatt, T. F. J. Pijpers, G. W. Harding, V. B. F. Mathot and H. Pasch, Macromolecules, 2012, 45, 2025–2034 CrossRef CAS.
- H. Tan, L. Li, Z. Chen, Y. Song and Q. Zheng, Polymer, 2005, 46, 3522–3527 CrossRef CAS PubMed.
- Y. Chen, Y. Chen, W. Chen and D. Yang, J. Appl. Polym. Sci., 2008, 108, 2379–2385 CrossRef CAS.
- J. Z. Liang and R. K. Y. Li, J. Appl. Polym. Sci., 2000, 77, 409–417 CrossRef CAS.
- A. van der Wal, R. Nijhof and R. J. Gaymans, Polymer, 1999, 40, 6031–6044 CrossRef CAS.
- H. Xu, Y. Zhang, J. Yang, L. Ye, Q. Wu, B. Qu, Q. Wang and Z. Wang, Polym. Chem., 2013, 4, 3028–3038 RSC.
- B. Na, W. Xu, R. Lv, Z. Li, N. Tian and S. Zou, Macromolecules, 2010, 43, 3911–3915 CrossRef CAS.
- Z. Li, S. Guo, W. Song and B. Hou, J. Mater. Sci., 2003, 38, 1793–1802 CrossRef CAS.
- J. Wang, C. Wang, X. Zhang, H. Wu and S. Guo, RSC Adv., 2014, 4, 20297–20307 RSC.
- S. Sinha Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539–1641 CrossRef PubMed.
- Y. Lin, H. Chen, C. M. Chan and J. Wu, Macromolecules, 2008, 41, 9204–9213 CrossRef CAS.
- H. Yang, Q. Zhang, M. Guo, C. Wang, R. Du and Q. Fu, Polymer, 2006, 47, 2106–2115 CrossRef CAS PubMed.
- H. Yang, X. Zhang, C. Qu, B. Li, L. Zhang, Q. Zhang and Q. Fu, Polymer, 2007, 48, 860–869 CrossRef CAS PubMed.
- Z. Bartczak, A. S. Argon, R. E. Cohen and M. Weinberg, Polymer, 1999, 40, 2347–2365 CrossRef CAS.
- Z. Bartczak, J. Macromol. Sci., Part B: Phys., 2002, 41, 1205–1229 CrossRef PubMed.
- Z. H. Liu, K. W. Kwok, R. K. Y. Li and C. L. Choy, Polymer, 2002, 43, 2501–2506 CrossRef CAS.
- C. M. Chan, J. Wu, J. X. Li and Y. K. Cheung, Polymer, 2002, 43, 2981–2992 CrossRef CAS.
- Y. S. Thio, A. S. Argon, R. E. Cohen and M. Weinberg, Polymer, 2002, 43, 3661–3674 CrossRef CAS.
- D. Shi, E. Liu, T. Tan, H. Shi, T. Jiang, Y. Yang, S. Luan, J. Yin, Y. W. Mai and R. K. Y. Li, RSC Adv., 2013, 3, 21563–21569 RSC.
- X. Zhou, J. Feng, J. Yi and L. Wang, Mater. Des., 2013, 49, 502–510 CrossRef CAS PubMed.
- R. Su, K. Wang, Q. Zhang, F. Chen, Q. Fu, W. Xu and B. Na, J. Phys. Chem. B, 2009, 113, 7423–7429 CrossRef CAS PubMed.
- T. Kurauchi and T. Ohta, J. Mater. Sci., 1984, 19, 1699–1709 CrossRef CAS.
- B. Yin, L. P. Li, Y. Zhou, L. Gong, M. B. Yang and B. H. Xie, Polymer, 2013, 54, 1938–1947 CrossRef CAS PubMed.
- L. P. Li, B. Yin, Y. Zhou, L. Gong, M. B. Yang, B. H. Xie and C. Chen, Polymer, 2012, 53, 3043–3051 CrossRef CAS PubMed.
- B. W. Qiu, F. Chen, Y. Lin, Y. G. Shangguan and Q. Zheng, Polymer, 2014, 55, 6176–6185 CrossRef CAS PubMed.
- C. H. Zhang, R. C. Chen, Y. G. Shangguan, Q. zheng and G. H. Hu, Chin. J. Polym. Sci., 2011, 29, 497–505 CrossRef CAS PubMed.
- H. Huo, S. Jiang, L. An and J. Feng, Macromolecules, 2004, 37, 2478–2483 CrossRef CAS.
- R. H. Somani, B. S. Hsiao, A. Nogales, H. Fruitwala, S. Srinivas and A. H. Tsou, Macromolecules, 2001, 34, 5902–5909 CrossRef CAS.
- A. M. C. Souza and N. R. Demarquette, Polymer, 2002, 43, 3959–3967 CrossRef CAS.
- C. Kock, M. Gahleitner, A. Schausberger and E. Ingolic, J. Appl. Polym. Sci., 2013, 128, 1484–1496 CAS.
- S. Wu, Polym. Eng. Sci., 1987, 27, 335–343 CAS.
- Y. J. Lin, V. Yakovleva, H. Y. Chen, A. Hiltner and E. Baer, J. Appl. Polym. Sci., 2009, 113, 1945–1952 CrossRef CAS.
- M. Hemmati, H. Nazokdast and H. Shariat Panahi, J. Appl. Polym. Sci., 2001, 82, 1129–1137 CrossRef CAS.
- K. A. Chaffin, F. S. Bates, P. Brant and G. M. Brown, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 108–121 CrossRef CAS.
- Z. Q. Fan, Y. Q. Zhang, J. T. Xu, H. T. Wang and L. X. Feng, Polymer, 2001, 42, 5559–5566 CrossRef CAS.
- L. Yang, R. H. Somani, I. Sics, B. S. Hsiao, R. Kolb, H. Fruitwala and C. Ong, Macromolecules, 2004, 37, 4845–4859 CrossRef CAS.
- J. Varga, J. Macromol. Sci., Part B: Phys., 2002, 41, 1121–1171 CrossRef PubMed.
- Q. Zheng, Y. Shangguan, S. K. Yan, Y. H. Song, M. Peng and Q. B. Zhang, Polymer, 2005, 46, 3163–3174 CrossRef CAS PubMed.
- C. Hedesiu, D. E. Demco, R. Kleppinger, A. A. Buda, B. Blümich, K. Remerie and V. M. Litvinov, Polymer, 2007, 48, 763–777 CrossRef CAS PubMed.
- G. R. Strobl and M. Schneider, J. Polym. Sci., Polym. Phys. Ed., 1980, 18, 1343–1359 CrossRef CAS.
- G. R. Strobl, M. J. Schneider and I. G. Voigtmartin, J. Polym. Sci., Polym. Phys. Ed., 1980, 18, 1361–1381 CrossRef CAS.
- H. Bai, F. Luo, T. Zhou, H. Deng, K. Wang and Q. Fu, Polymer, 2011, 52, 2351–2360 CrossRef CAS PubMed.
- K. Yamada, M. Hikosaka, A. Toda, S. Yamazaki and K. Tagashira, Macromolecules, 2003, 36, 4790–4801 CrossRef CAS.
- G. W. H. Höhne, Polymer, 2002, 43, 4689–4698 CrossRef.
- A. T. Lorenzo, M. L. Arnal, A. J. Müller, M. C. Lin and H. L. Chen, Macromol. Chem. Phys., 2011, 212, 2009–2016 CrossRef CAS.
- B. W. Qiu, F. Chen, Y. G. Shangguan, Y. Lin and Q. Zheng, Chin. J. Polym. Sci., 2015 DOI:10.1007/s10118-015-1556-8.
- J. Rösch, Polym. Eng. Sci., 1995, 35, 1917–1922 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |