
Design and synthesis of pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones with C-3′ side chains as potent Met kinase inhibitors†
Chia-Wei Liua,
Chun-Liang Laia,
Yu-Hsiang Lina,
Li-Wei Tenga,
Sheng-chuan Yanga,
Win-Yin Weia,
Shu Fu Lina,
Ju-Ying Yanga,
Hung-Jyun Huanga,
Ru-Wen Wanga,
Chao-Cheng Chianga,
Mei-Hui Leea,
Yu-Chuan Wangb,
Shih-Hsien Chuanga,
Jia-Ming Changa,
Ying-Shuan E. Leea and
Jiann-Jyh Huang*ab
aDevelopment Center for Biotechnology, No. 101, Lane 169, Kangning St., Xizhi District, New Taipei City 22180, Taiwan. E-mail: lukehuang@mail.ncyu.edu.tw; Fax: +886 5 271 7901; Tel: +886 5 271 7959
bDepartment of Applied Chemistry, National Chiayi University, No. 300, Syuefu Rd., Chiayi City 60004, Taiwan
First published on 30th October 2014
Abstract
Pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones of scaffold 4 with various C-3′ side chains were designed as potent Met kinase inhibitors. Structural optimization led to compounds 10, 20, and 22–24 which demonstrated subnanomolar IC50 values in the biochemical assay. The potent compound 20 inhibited Met with IC50 value of 0.37 nM and the proliferation of MKN45 cells with IC50 of 0.22 μM. It suppressed Met autophosphorylation with the downstream signaling through Gab-1, PLC-γ, FAK, Akt, STAT3, and ERK in cell. Complete inhibition of STAT3 and ERK phosphorylation was observed in MKN45 cells treated with 20 at the concentration of 100 nM. A computation simulation study was performed to reveal the interaction of 20 with Met.
Introduction
Met is a transmembrane receptor tyrosine kinase expressed in normal and malignant cells. Upon binding to its natural ligand hepatocyte growth factor/scatter factor (HGF/SF), Met induces several signaling pathways responsible for cell proliferation, motility, migration, and survival.1–3 The cellular events play an important role in normal embryonic development and wound healing4 but nevertheless, overexpression of HGF and/or Met or activating mutation of Met can lead to cancer.5 As a result, Met is considered as a potential target for cancer therapy.6–8C-5 substituted pyrrole–indolin-2-ones SU11274 (1)9 and PHA665752 (2)10 are the prototype Met kinase inhibitors from in silico design before the three dimensional structure of Met was characterized (Fig. 1). They are obtained using the homology model of Met built from the closely related FGFR1 kinase.11 The C-4′ amino–amido side chains of 1 and 2 were designed to modulate the pharmacokinetic properties12 and would provide additional interactions with the enzyme to enhance the activities. Though 1 and 2 are only used as the biology tool, subsequent scaffold hopping based on the structure of 2 led to PF-2341066 (3),13 the most advanced Met and ALK kinase inhibitor recently approved by FDA for the treatment of NSCLC (see Fig. 1).
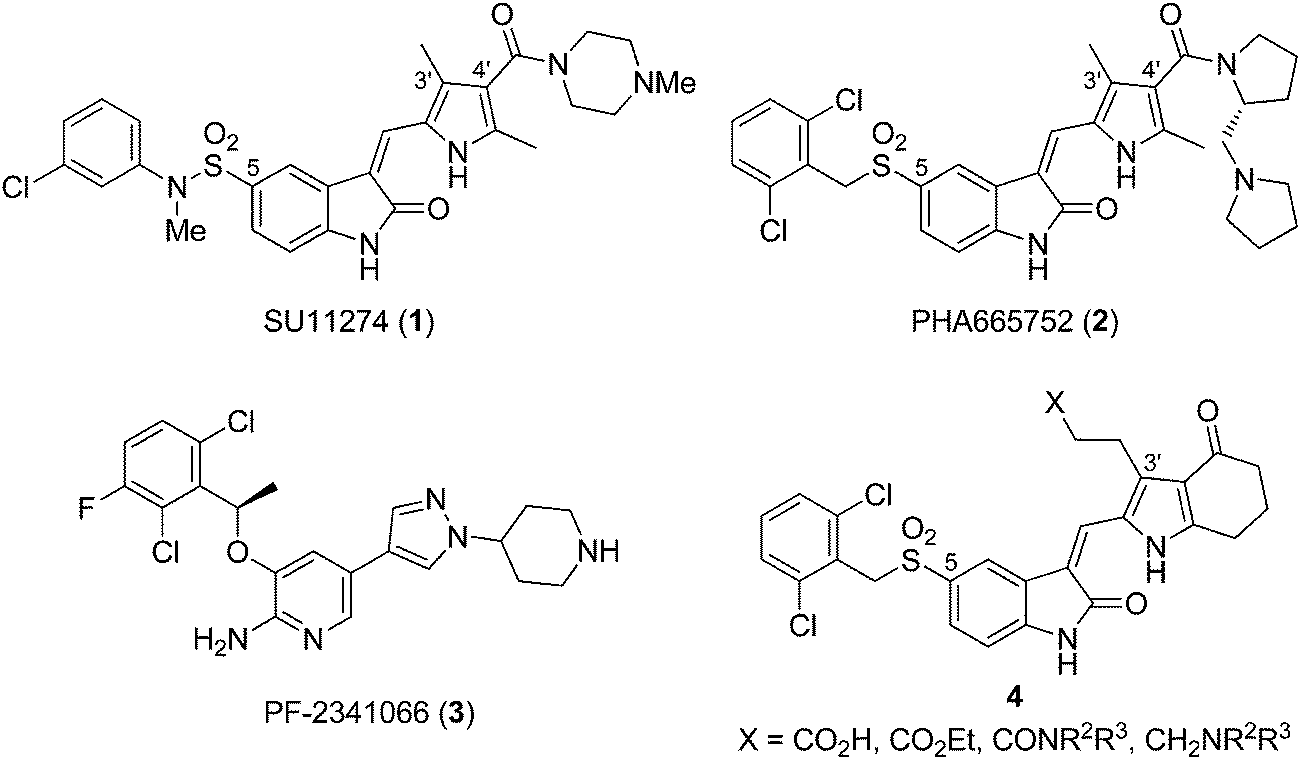 | ||
| Fig. 1 Structures of SU11274 (1), PHA665752 (2), PF-2341066 (3), and pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-one of scaffold 4. | ||
Based on the literatures reporting the discovery of pyrrole–indolin-2-ones as multikinase inhibitors for VEGFR-2, FGFR1, and PDGFR-β kinases, different inhibition profiles are observed with different positions of the side chains.14,15 For FGFR1 inhibition, moving the C-4′ side chain in a pyrrole–indolin-2-one to the C-3′ position provided compounds with 5- to 10-fold increased potency (see the structures of 1 and 2 in Fig. 1). As compound 2 was designed from the Met homology model of FGFR1, we envisaged that pyrrole–indolin-2-ones with C-3′ side chains would also be potent Met inhibitors. Currently, the role of C-3′ side chain for Met inhibition has not been studied in detail.
In this study, we used pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-one of scaffold 4 as the template to study the role of C-3′ substituents for the inhibition of Met kinase. Except for the C-3′ substituents, scaffold 4 possesses a pyrrole-fused cyclohexanone moiety for patent purposes.16 Subsequent optimization led to compounds 10, 20, and 22–24 that demonstrated subnanomolar IC50 values for Met inhibition in the biochemical assay. A computation modeling was used to reveal the different interactions of the C-3′ side chain of 4 with Met in comparison with the C-4′ side chain of 2. We expected the structural information would contribute to the further design of new scaffolds against Met, as in the case of designing 3 from 1 and 2.
Results and discussion
Chemistry
Scheme 1 presents the synthesis of pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones 7–12 of scaffold 4 bearing C-3′ carboxyl-, ethoxycarbonyl-, and amido-ethyl side chains (X = CO2H, CO2Et, and CONR2R3 in 4, Fig. 1). 2-Formylpyrrole 5, prepared according to our published procedures,17 was condensed with 5-[(2,6-dichlorobenzyl)sulfonyl]indolin-2-one (6) to provide compound 7 bearing a C-3′ carboxylethyl group in 89% yield. Esterification of 7 in EtOH in the presence of SOCl2 provided 8 that possessed a C-3′ ethoxycarbonylethyl group in 92% yield. Amidation of the carboxyl group in 7 by various amines using CDI as the coupling agent provided pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones 9–12 bearing a C-3′ amido-ethyl group in 70–85% yields.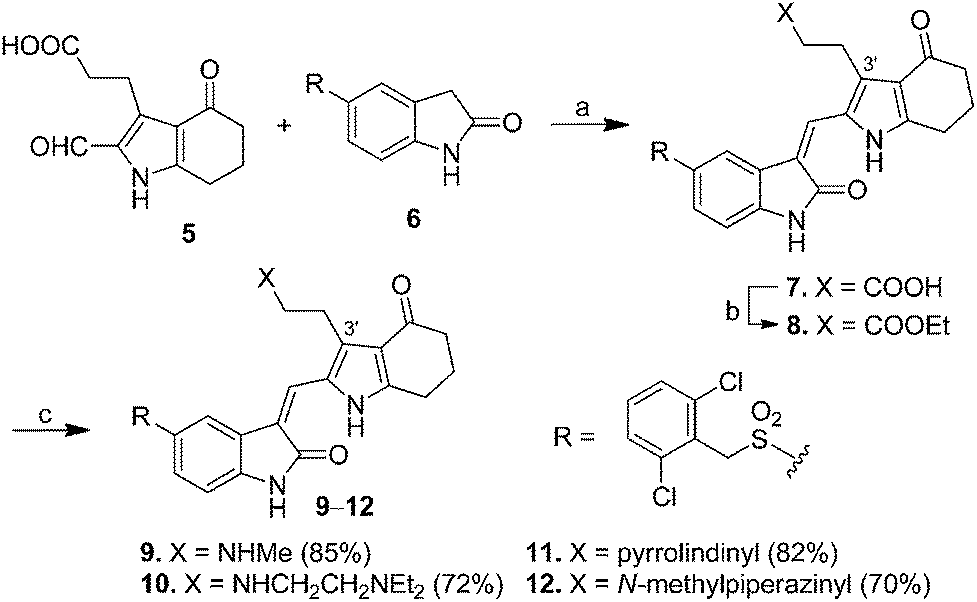 | ||
| Scheme 1 Synthesis of pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones 7–12 bearing C-3′ carboxyl-, ethoxycarbonyl-, or amido-ethyl side chains as Met kinase inhibitors. Reagents and conditions: (a) piperidine, EtOH, reflux, 12 h, 89%; (b) SOCl2, EtOH, 92%; and (c) 7, amine, CDI, CH2Cl2, 70–85%. | ||
For the synthesis of pyrrole–indolin-2-ones 20–24 bearing C-3′ aminopropyl side chains (X = CH2NR2R3 in 4, Fig. 1), pyrroles 13a–e served as the starting material to react with phenylsulfonyl chloride (PhSO2Cl) in the presence of NaOH to provide the N1-protected pyrrole 14a–e in 86–94% yields (Scheme 2). The carbonyl groups in 14a–e could be reacted with ethane-1,2-dithiol to give the dithiane 15a–e in 87–99% yields. Removal of phenylsulfonyl group in 15a–e under basic condition provided 16a–e in 96% to >99% yields, which was treated with LiAlH4 in THF at room temperature to afford amine 17a–e in 60–96% yields. Introduction of a formyl group to the C-2 position of 17a–e was accomplished by use of Vilsmeier–Haack reaction to generate 18a–e in 71–97% yields. Deprotection of the dithianyl group using HgCl2 afforded 19a–e in 66–93% yields, which was eventually condensed with 5-(2,6-dichlorobenzylsulfonyl)indolin-2-one (6) to provide the corresponding 20–24 with aminopropyl groups at their C-3′ positions in 60–78% yields.
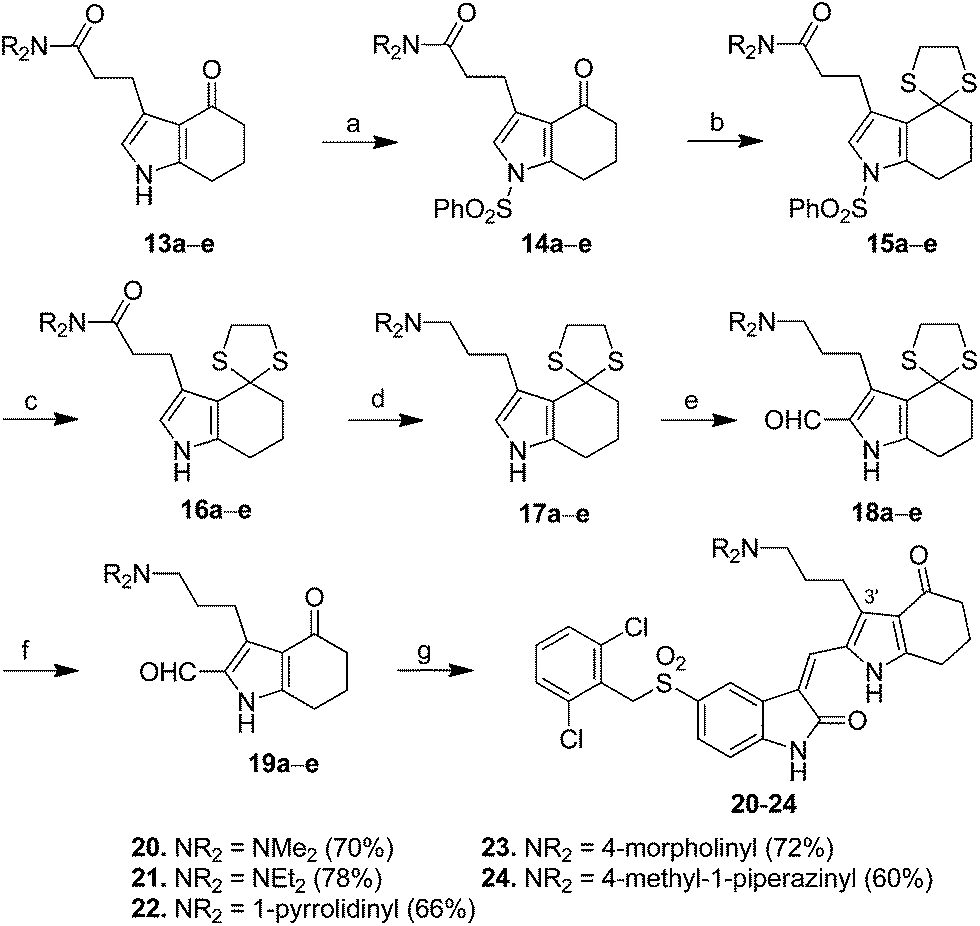 | ||
| Scheme 2 Synthesis of pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones 20–24 equipped with C-3′ aminopropyl side chain. Reagents and conditions: (a) PhSO2Cl, NaOH, CH2Cl2, 86–94%; (b) ethane-1,2-dithiol, BF3·OEt2, MeOH, 87–99%; (c) NaOH, H2O, MeOH, reflux, 96% to >99%; (d) LiAlH4, THF, r.t., 60–96%; (e) POCl3, DMF, 71–97%; (f) HgCl2, CaCO3, CH3CN, H2O, 66–93%; and (g) 5-(2,6-dichlorobenzylsulfonyl)indolin-2-one (6), piperidine, EtOH, reflux, 60–78%. | ||
In vitro potency
The IC50 values of pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones 7–12 and 20–24 for the inhibition of Met kinase and the proliferation of MKN45 cells (gastric carcinoma) that carry an amplified Met proto-oncogene18 are presented in Table 1. We also tested the potencies of 7–12 and 20–24 for the inhibition of Aurora kinases for comparison due to pyrrole–indolin-2-ones possessing a C-5 bulky group and a C-3 carboxylethyl side chain were found to show Aurora kinase inhibitory activities in our previous study.17 As shown in Table 1, the prototype pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-one 7 bearing a C-3′ carboxyethyl group displayed an IC50 value of 30.40 nM for Met inhibition. However, it did not show significant antiproliferative activity for MKN45 cells (IC50 > 10 μM). Compound 8, the ethyl ester analog to 7, showed a slightly improved potency (IC50: 20.20 nM) as well as enhanced antiproliferative activity for MKN45 (IC50: 3.25 μM). These two compounds did not significantly inhibit Aurora kinases expect 7 inhibited Aurora A with an IC50 value of 855 nM.
|
|||||
|---|---|---|---|---|---|
| Compd | X | Met IC50a (nM) | MKN45 IC50a,b (μM) | % inhibition at 1.0 μM | |
| Aurora A | Aurora B | ||||
| a The IC50 values were averaged from two independent dose–response curves.b A 72 h cell proliferation assay.c IC50: 855 nM.d Obtained from literature.19 | |||||
| 7 | 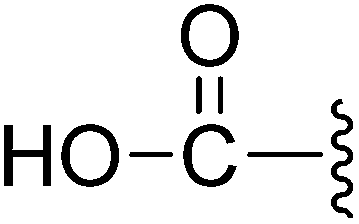 |
30.40 | >10 | 65.5%c | 9.4% |
| 8 | 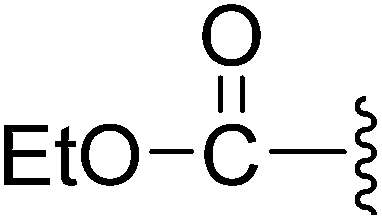 |
20.20 | 3.25 ± 0.41 | 22.7% | 16.3% |
| 9 | 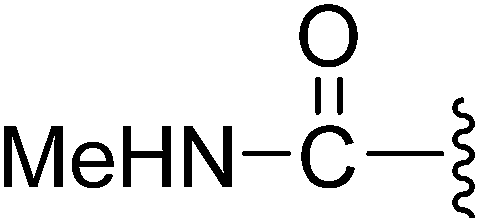 |
4.22 | >10 | 13.0% | 6.9% |
| 10 | 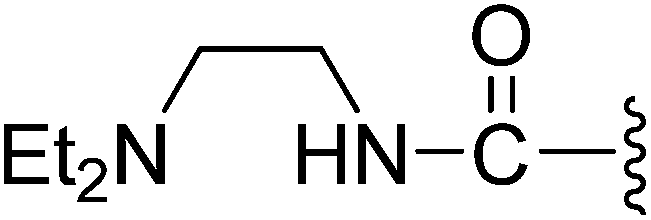 |
0.15 | 2.34 ± 0.13 | 10.8% | 9.2% |
| 11 | 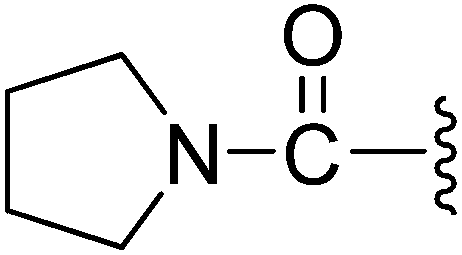 |
7.17 | 9.25 ± 0.32 | 2.2% | 5.5% |
| 12 | 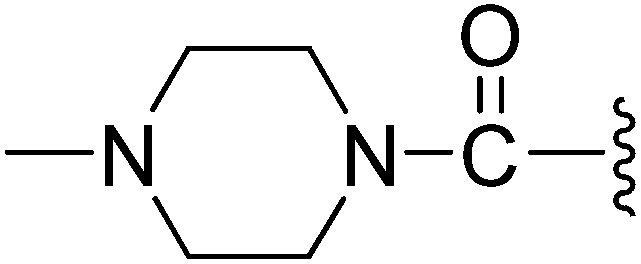 |
2.94 | 0.99 ± 0.10 | 8.7% | 1.3% |
| 20 |  |
0.37 | 0.22 ± 0.03 | 2.7% | 34.3% |
| 21 | 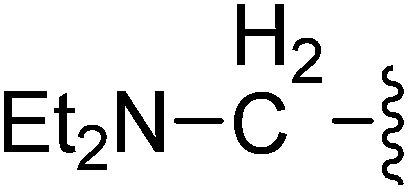 |
7.70 | 0.40 ± 0.05 | 1.9% | 6.2% |
| 22 | 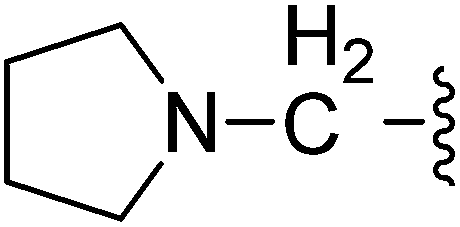 |
0.31 | 0.82 ± 0.08 | 4.2% | 22.3% |
| 23 | 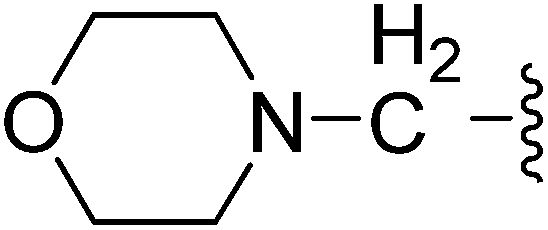 |
0.43 | 0.47 ± 0.05 | 23.7% | 35.4% |
| 24 | 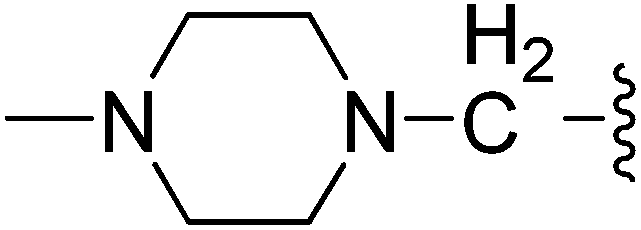 |
0.26 | 0.24 ± 0.02 | 6.0% | 15.3% |
| 2 | — | 9d | 0.10 ± 0.03 | — | — |
For further improving the Met and MKN45 inhibitory activities, we evaluated compounds 9–12 with the C-3′ amidoethyl substituents. These compounds showed remarkable improvements for Met inhibition with IC50 values less than 10 nM (IC50 for 2: 9 nM).19 The most potent amido compound 10 with a N,N-diethylaminoethylamino (Et2NCH2CH2NH–) substituent inhibited Met at subnanomolar concentration (IC50: 0.15 nM) and showed antiproliferative activity for MKN45 cells at low micromolar concentration (IC50: 2.34 μM). The N-methylpiperazinyl analog 12 displayed the most potent antiproliferative activity (MKN45 IC50: 0.99 μM) among the amido compounds 9–12. Respective comparison of kinase and cell activities of 9 with 11 and 10 with 12 suggested that the NH group in the amido moiety would profit the enzymatic but not the cell activity. The difference in the cell activities of these compounds might come from their different cell permeability or inhibition profiles against other kinases, e.g., VEGFR-2, PDGFR, and FGFR1.15
Pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones 20–24 bound with C-3′ aminopropyl groups showed enhanced activities both for Met kinase and MKN45 cells (Table 1). Except for 21 with a diethylamino group, these compounds inhibited Met at subnanomolar concentrations (IC50: 0.26–0.43 nM) and demonstrated better antiproliferative activities than their carboxyl, ester, and amido analogs (i.e. 7–12) on MKN45 cells with IC50 values less than 1.0 μM. The promising results from the biochemical assay shown in Table 1 indicated that pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones of scaffold 4 with C-3′ side chains also showed promising Met kinase inhibitory activity as their C-4′ analog 2. Furthermore, the use of the C-3′ ethyl ester, amido, and amino side chains also diminished the activity of the corresponding compounds against Aurora A and Aurora B kinases (IC50 > 1.0 μM, Table 1) in comparison with the use of a C-3′ carboxyl side chain (compound 7). A great selectivity (∼100- to 1000-fold) was thus achieved for Met over Aurora kinases for the compounds in Table 1.
Western blotting
As shown in Table 1, the great enhancement for the Met inhibition by compounds 20 and 22–24 did not reflect on their antiproliferative activities (MKN45 IC50: 0.22–0.82 μM). We then characterized the effect of potent compound 20 (MKN45 IC50: 0.22 μM) on Met signal transduction in MKN45 cells by Western blot, and the results are presented in Fig. 2. Strong inhibition of tyrosine phosphorylation in the Met kinase domain (pY1234/1235) was observed with 20 at a concentration of 1.1 μM and the reduced level of phosphorylated Met (IC50 ∼ 0.1–1.1 μM) correlated to the antiproliferative IC50 value of 20 (0.22 μM). The phosphorylation of Met docking protein Gab-1 was completely inhibited at the concentration of 0.4 μM. Compound 20 also potently inhibited the constitutive downstream signaling through PLC-γ, FAK, Akt, STAT-3, and Erk, which provided the evidence of Met inhibition by 20 in cell. Regarded as the downstream cellular effectors of Met inhibition, the phosphorylation of STAT3 and ERK in MKN45 cells was completely abolished by 20 at the concentration below 100 nM, showing better correlation to the results from biochemical assay than the antiproliferative activity.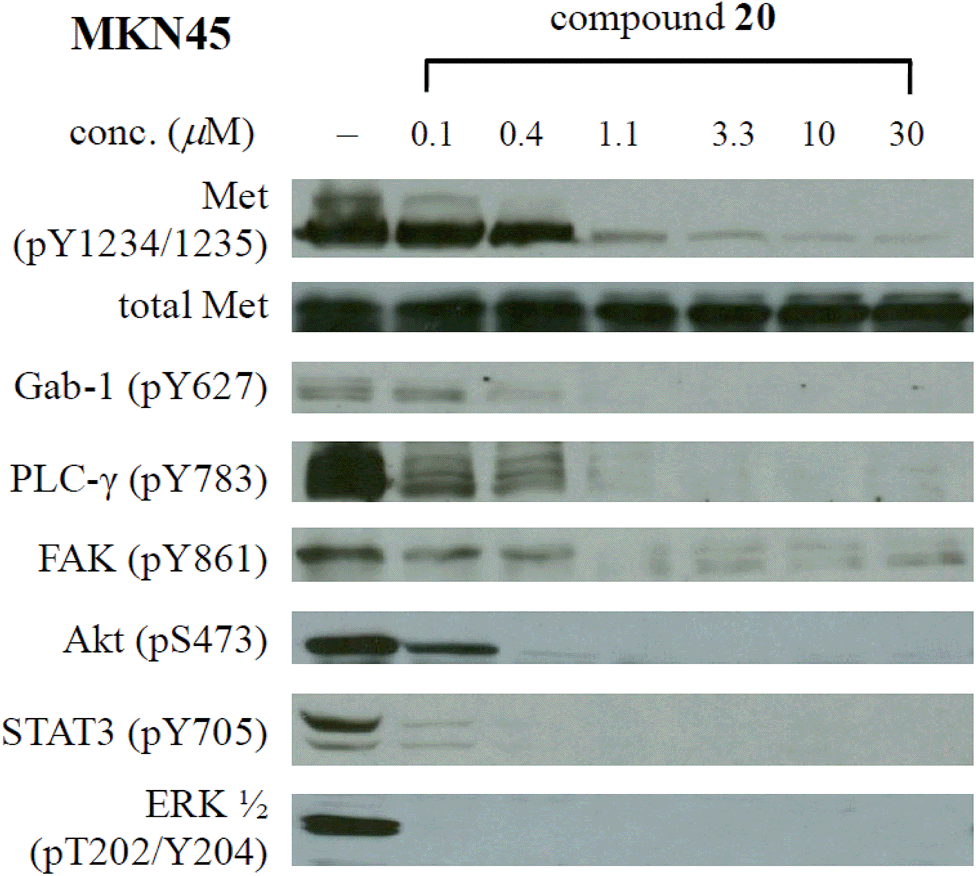 | ||
| Fig. 2 Effect of 20 on Met phosphorylation and signal transduction in MKN45 cells. | ||
Kinase profiling
Since compound 20 shared a similar scaffold with the multikinase inhibitor sunitinib,14 we explored its inhibition profile against a panel of 451 kinases at the concentration of 1.0 μM on DiscoveRx Kinomescan scanMax platform.20 The results revealed that CAMKK1, CDKL2, CSF1R, DCAMKL3, FLT3 (D835Y), FLT3 (ITD), GRK1, GRK7, IRAK4, JAK2 (JH1domain-catalytic), MAP4K2, MAP4K3, MAP4K4, MAP4K5, MET, MET(M1250T), MET(Y1235D), MST2, PDGFRB, RSK3 (Kin.Dom.1-N-terminal), TAOK1, TAOK2, TAOK3, TNIK, TRKB, and YSK4 kinases were inhibited significantly by 20 with a percentage of control of <10% (see the table in the ESI†).Molecular modeling
To explore the interaction of pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones of scaffold 4 with Met, we modeled the binding mode of the potent compound 20 as well as reference compound 2 in Met (PDB code 2RFS)12 using docking simulations. After energy minimization, the binding poses of 20 and 2 were overlapped as illustrated in Fig. 3. Obviously, 20 shared similar binding pose to 2 in Met. Both compounds formed two H-bonds from N1–H and N1′–H with Pro1158 and Met1160, respectively, and an H-bond from C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O with Met1160 in the hinge region. The oxygen atom of the C-5 sulfonyl group formed an H-bond with Asp1222, and the 2,6-dichlorophenyl fitted the hydrophobic pocket formed by Val1092, Tyr1230, and Gly1085 from the analysis of Ligplot.21 However, the side chain at the C-3′ position of 20 headed to the different region from the C-4′ amino–amido group in 2, and thus interacted with different residues in Met other than 2: 20 interacted with Ile1084 and 2 interacted with Tyr1159 and His1094 (Fig. 3). The hydrophilic interaction of the C-3′ dimethylamino group might contribute the promising activity of 20.
O with Met1160 in the hinge region. The oxygen atom of the C-5 sulfonyl group formed an H-bond with Asp1222, and the 2,6-dichlorophenyl fitted the hydrophobic pocket formed by Val1092, Tyr1230, and Gly1085 from the analysis of Ligplot.21 However, the side chain at the C-3′ position of 20 headed to the different region from the C-4′ amino–amido group in 2, and thus interacted with different residues in Met other than 2: 20 interacted with Ile1084 and 2 interacted with Tyr1159 and His1094 (Fig. 3). The hydrophilic interaction of the C-3′ dimethylamino group might contribute the promising activity of 20.
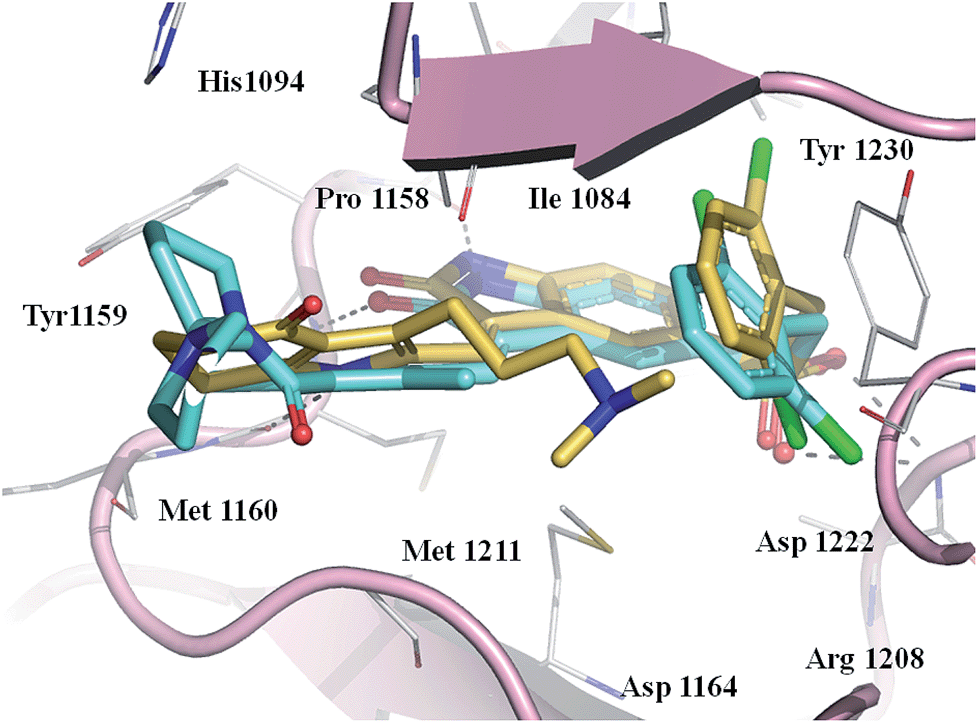 | ||
| Fig. 3 Overlapping of the docked binding modes of compounds 2 and 20 in the ATP binding site of Met kinase. Compound 2 is shown as light blue sticks, 20 is shown as yellow sticks, hydrogen bonds are shown as dashed gray line, and the oxygen, nitrogen, and sulfur atoms are colored in red, blue, and orange, respectively. The C-3′ side chain of 20 faced the region around Ile1084 and the C-4′ side chain of 2 faced the region around Tyr1159 and His1094. | ||
Conclusions
Pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones of scaffold 4 with a C-3′ side chain was designed as potent Met kinase inhibitors. Structural optimization led to compounds 10, 20, and 22–24 that showed subnanomolar IC50 values in the biochemical assay. Computation modeling revealed that the C-3′ side chain of 4 interacted with Ile1084 and would contribute to its promising activity. The potent compound 20 possessed an IC50 value of 0.37 nM also inhibited the proliferation of MKN45 cells (IC50: 0.22 μM). Cellular events including the inhibition of Met autophosphorylation and the downstream signaling through Gab-1, PLC-γ, FAK, Akt, STAT-3, and ERK were found on MKN45 cells treated with 20. Among the proteins, STAT3 and ERK were completely inhibited by 20 at the concentration of 100 nM.Experimental
Chemistry
Reagents were used as purchased without further purification. 5-(2,6-Dichlorobenzyl)sulfonylindolin-2-one (6) was prepared according to literature procedures.22,23 Proton NMR spectra were recorded on a Bruker (500 MHz) spectrometer with CDCl3 or DMSO-d6 as solvent. Multiplicities are abbreviated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; J, coupling constant (hertz). Carbon-13 NMR spectra were obtained from a Bruker spectrometer (125 MHz) by use of CDCl3 or DMSO-d6 as solvent. Carbon-13 chemical shifts are referenced to the center peak of CDCl3 (δ 77.0 ppm) or DMSO-d6 (δ 39.5 ppm). ESI-MS spectra were recorded with an Applied Biosystems API 300 mass spectrometer. The purities of the compounds were greater than 95% as determined from reversed-phase HPLC. The synthesis of compounds 5, 13a–e, 17a–e, 18a–e, and 19a–e was followed our previously reported procedures.17N,N-Dimethyl-3-[4-oxo-1-(phenylsulfonyl)-4,5,6,7-tetrahydro-1H-indol-3-yl]propanamide (14a). Yield: 94%; 1H NMR (CDCl3) δ 7.85 (d, J = 7.6 Hz, 2H), 7.66 (t, J = 7.5 Hz, 1H), 7.55–7.58 (m, 2H), 7.06 (s, 1H), 3.02 (s, 3H), 2.89–2.98 (m, 7H), 2.56–2.61 (m, 2H), 2.36–2.41 (m, 2H), 2.02–2.09 (m, 2H); ESI-MS m/z 374.7 (M + H)+.
N,N-Diethyl-3-[4-oxo-1-(phenylsulfonyl)-4,5,6,7-tetrahydro-1H-indol-3-yl]propanamide (14b). Yield: 93%; 1H NMR (CDCl3) δ 7.85 (d, J = 7.6 Hz, 2H), 7.66 (t, J = 7.5 Hz, 1H), 7.54–7.57 (m, 2H), 7.06 (s, 1H), 3.29–3.38 (m, 4H), 2.96 (t, J = 6.8 Hz, 4H), 2.54–2.59 (m, 2H), 2.37–2.43 (m, 2H), 2.03–2.10 (m, 2H), 1.07–1.13 (m, 6H); ESI-MS m/z 402.9 (M + H)+.
3-[3-Oxo-3-(pyrrolidin-1-yl)propyl]-1-(phenylsulfonyl)-6,7-dihydro-1H-indol-4(5H)-one (14c). Yield: 89%; 1H NMR (CDCl3) δ 7.85 (d, J = 7.5 Hz, 2H), 7.65 (t, J = 7.5 Hz, 1H), 7.53–7.56 (m, 2H), 7.06 (s, 1H), 3.38–3.45 (m, 4H), 2.92–2.97 (m, 4H), 2.50–2.55 (m, 2H), 2.34–2.41 (m, 2H), 2.02–2.09 (m, 2H), 1.85–1.92 (m, 2H), 1.77–1.84 (m, 2H); ESI-MS m/z 400.8 (M + H)+.
3-(3-Morpholino-3-oxopropyl)-1-(phenylsulfonyl)-6,7-dihydro-1H-indol-4(5H)-one (14d). Yield: 89%; 1H NMR (CDCl3) δ (d, J = 7.6 Hz, 2H), 7.67 (t, J = 7.5 Hz, 1H), 7.56–7.59 (m, 2H), 7.07 (s, 1H), 3.55–3.65 (m, 8H), 2.90–2.99 (m, 4H), 2.57–2.62 (m, 2H), 2.37–2.42 (m, 2H), 2.04–2.11 (m, 2H); ESI-MS m/z 417.0 (M + H)+.
3-[3-(4-Methylpiperazin-1-yl)-3-oxopropyl]-1-(phenylsulfonyl)-6,7-dihydro-1H-indol-4(5H)-one (14e). Yield: 86%; 1H NMR (CDCl3) δ 7.86 (d, J = 7.5 Hz, 2H), 7.67 (t, J = 7.5 Hz, 1H), 7.56–7.59 (m, 2H), 7.06 (s, 1H), 3.50–3.64 (m, 4H), 2.90–2.98 (m, 4H), 2.57–2.62 (m, 2H), 2.33–2.41 (m, 6H), 2.28 (s, 3H), 2.04–2.10 (m, 2H); ESI-MS: m/z 430.0 (M + H)+.
N,N-Dimethyl-3-[1′-(phenylsulfonyl)-1′,5′,6′,7′-tetrahydrospiro[[1,3]dithiolane-2,4′-indole]-3′-yl]propanamide (15a). Yield: 87%; 1H NMR (CDCl3) δ 7.77 (d, J = 7.5 Hz, 2H), 7.62 (t, J = 7.5 Hz, 1H), 7.51–7.54 (m, 2H), 6.99 (s, 1H), 3.42–3.50 (m, 2H), 3.30–3.36 (m, 2H), 3.06–3.11 (m, 2H), 3.02 (s, 3H), 2.97 (s, 3H), 2.61–2.70 (m, 4H), 2.15–2.19 (m, 2H), 1.80–1.89 (m, 2H); ESI-MS m/z 450.8 (M + H)+.
N,N-Diethyl-3-[1′-(phenylsulfonyl)-1′,5′,6′,7′-tetrahydrospiro[[1,3]dithiolane-2,4′-indole]-3′-yl]propanamide (15b). Yield: 92%; 1H NMR (CDCl3) δ 7.77 (d, J = 7.6 Hz, 2H), 7.61 (t, J = 7.4 Hz, 1H), 7.50–7.54 (m, 2H), 6.99 (s, 1H), 3.44–3.51 (m, 2H), 3.37–3.43 (m, 2H), 3.30–3.36 (m, 4H), 3.07–3.13 (m, 2H), 2.61–2.69 (m, 4H), 2.15–2.19 (m, 2H), 1.82–1.88 (m, 2H), 1.10–1.20 (m, 6H); ESI-MS m/z 478.8 (M + H)+.
3-[1′-(Phenylsulfonyl)-1′,5′,6′,7′-tetrahydrospiro[[1,3]dithiolane-2,4′-indole]-3′-yl]-1-(pyrrolidin-1-yl)propan-1-one (15c). Yield: 90%; 1H NMR (CDCl3) δ 7.74–7.80 (m, 2H), 7.62 (t, J = 7.5 Hz, 1H), 7.51–7.54 (m, 2H), 6.99 (s, 1H), 3.39–3.53 (m, 6H), 3.28–3.38 (m, 2H), 3.05–3.15 (m, 2H), 2.57–2.68 (m, 4H), 2.12–2.22 (m, 2H), 1.92–2.00 (m, 2H), 1.79–1.90 (m, 4H); ESI-MS m/z 477.0 (M + H)+.
1-Morpholino-3-[1′-(phenylsulfonyl)-1′,5′,6′,7′-tetrahydrospiro[[1,3]dithiolane-2,4′-indole]-3′-yl]propan-1-one (15d). Yield: 99%; 1H NMR (CDCl3) δ 7.77 (d, J = 7.4 Hz, 2H), 7.62 (t, J = 7.4 Hz, 1H), 7.51–7.54 (m, 2H), 6.99 (s, 1H), 3.61–3.69 (m, 6H), 3.42–3.52 (m, 4H), 3.31–3.38 (m, 2H), 3.07–3.12 (m, 2H), 2.61–2.71 (m, 4H), 2.15–2.19 (m, 2H), 1.83–1.87 (m, 2H); ESI-MS m/z 492.9 (M + H)+.
1-(4-Methylpiperazin-1-yl)-3-[1′-(phenylsulfonyl)-1′,5′,6′,7′-tetrahydrospiro[[1,3]dithiolane-2,4′-indole]-3′-yl]propan-1-one (15e). Yield: 96%; 1H NMR (CDCl3) δ 7.77 (d, J = 7.5 Hz, 2H), 7.62 (t, J = 7.5 Hz, 1H), 7.50–7.53 (m, 2H), 6.99 (s, 1H), 3.64–3.68 (m, 2H), 3.50–3.54 (m, 2H), 3.42–3.49 (m, 2H), 3.30–3.37 (m, 2H), 3.07–3.12 (m, 2H), 2.61–2.72 (m, 4H), 2.36–2.40 (m, 4H), 2.31 (s, 3H), 2.16–2.20 (m, 2H), 1.82–1.88 (m, 2H); ESI-MS m/z 506.0 (M + H)+.
(Z)-3-(2-{[5-(2,6-Dichlorobenzylsulfonyl)-2-oxoindolin-3-ylidene]methyl}-4-oxo-4,5,6,7-tetrahydro-1H-indol-3-yl)propanoic acid (7). Yield: 89%; 1H NMR (DMSO-d6) δ 13.65 (s, 1H), 12.11 (brs, 1H), 11.52 (s, 1H), 8.21 (d, J = 1.6 Hz, 1H), 7.94 (s, 1H), 7.46–7.52 (m, 3H), 7.39 (dd, J = 8.7, 7.4 Hz, 1H), 7.06 (d, J = 8.2 Hz, 1H), 4.88 (s, 2H), 3.20 (t, J = 7.6 Hz, 2H), 2.93 (t, J = 6.1 Hz, 2H), 2.52 (t, J = 7.6 Hz, 2H), 2.40–2.44 (m, 2H), 2.02–2.10 (m, 2H); 13C NMR (DMSO-d6) δ 19.28, 21.83, 21.91, 33.87, 37.50, 56.89, 108.78, 114.70, 118.27, 118.77, 124.91, 124.99, 125.43, 126.16, 126.53, 127.81, 130.16, 131.02, 133.03, 135.51, 142.09, 147.59, 168.70, 172.97, 192.83; ESI-MS m/z 573.0 (M + H)+.
(Z)-5-(2,6-Dichlorobenzylsulfonyl)-3-({3-[3-(dimethylamino)propyl]-4-oxo-4,5,6,7-tetrahydro-1H-indol-2-yl}methylene)indolin-2-one (20). Yield: 70%; 1H NMR (DMSO-d6) δ 13.51 (s, 1H), 11.46 (brs, 1H), 7.97 (s, 1H), 7.42–7.57 (m, 3H), 7.34–7.42 (m, 2H), 7.07 (d, J = 6.9 Hz, 1H), 4.87 (s, 2H), 2.92–3.05 (m, 4H), 2.35–2.45 (m, 4H), 1.97–2.19 (m, 8H), 1.57–1.72 (m, 2H); 13C NMR (DMSO-d6) δ 19.73, 21.84, 21.96, 26.58, 37.56, 43.98, 56.37, 56.86, 108.88, 114.08, 117.69, 118.80, 124.90, 124.95, 125.04, 125.77, 126.62, 127.83, 130.17, 130.82, 134.51, 135.52, 142.03, 147.73, 168.67, 192.72; ESI-MS m/z 586.0 (M + H)+.
(Z)-5-(2,6-Dichlorobenzylsulfonyl)-3-({3-[3-(diethylamino)propyl]-4-oxo-4,5,6,7-tetrahydro-1H-indol-2-yl}methylene)indolin-2-one (21). Yield: 78%; 1H NMR (DMSO-d6) δ 13.62 (s, 1H), 8.11 (s, 1H), 7.89 (s, 1H), 7.43–7.56 (m, 3H), 7.37–7.42 (m, 1H), 7.07 (d, J = 8.10 Hz, 1H), 4.87 (s, 2H), 2.90–3.02 (m, 4H), 2.32–2.47 (m, 8H), 2.01–2.12 (m, 2H), 1.65–1.71 (m, 2H), 0.91 (t, J = 7.1 Hz, 6H); 13C NMR (DMSO-d6) δ 10.36, 21.02, 21.85, 21.98, 26.59, 37.59, 44.85, 50.24, 56.88, 108.86, 114.10, 117.77, 118.83, 124.97, 125.12, 125.48, 126.33, 126.51, 127.81, 130.15, 130.95, 134.42, 135.50, 142.04, 147.72, 168.70, 192.67; ESI-MS m/z 614.0 (M + H)+.
(Z)-5-(2,6-Dichlorobenzylsulfonyl)-3-({4-oxo-3-[3-(pyrrolidin-1-yl)propyl]-4,5,6,7-tetrahydro-1H-indol-2-yl}methylene)indolin-2-one (22). Yield: 66%; 1H NMR (DMSO-d6) δ 13.62 (s, 1H), 11.52 (s, 1H), 8.05 (s, 1H), 7.86 (s, 1H), 7.45–7.50 (m, 3H), 7.34–7.41 (m, 1H), 7.05 (d, J = 8.2 Hz, 1H), 4.85 (s, 2H), 3.03 (t, J = 6.8 Hz, 2H), 2.93 (t, J = 5.9 Hz, 2H), 2.27–2.42 (m, 8H), 2.01–2.09 (m, 2H), 1.63–1.73 (m, 6H); 13C NMR (DMSO-d6) δ 20.92, 21.84, 21.97, 22.17, 28.05, 37.58, 52.45, 53.26, 56.87, 108.87, 114.18, 117.66, 118.78, 124.93, 124.99, 125.04, 126.40, 126.51, 127.81, 130.17, 130.93, 134.71, 135.52, 142.03, 147.70, 168.70, 192.70; ESI-MS m/z 612.0 (M + H)+.
(Z)-5-(2,6-Dichlorobenzylsulfonyl)-3-{[3-(3-morpholinopropyl)-4-oxo-4,5,6,7-tetrahydro-1H-indol-2-yl]methylene}indolin-2-one (23). Yield: 72%; 1H NMR (DMSO-d6) δ 13.64 (s, 1H), 11.48 (brs, 1H), 8.21 (s, 1H), 7.88 (s, 1H), 7.46–7.50 (m, 3H), 7.33–7.41 (m, 1H), 7.05 (d, J = 8.2 Hz, 1H), 4.86 (s, 2H), 3.51–3.57 (m, 4H), 3.02 (t, J = 7.1 Hz, 2H), 2.93 (t, J = 6.0 Hz, 2H), 2.21–2.42 (m, 8H), 1.99–2.09 (m, 2H), 1.67–1.74 (m, 2H); 13C NMR (DMSO-d6) δ 21.12, 21.84, 21.98, 26.07, 37.58, 52.34, 56.35, 56.90, 65.22, 108.81, 114.20, 118.05, 118.84, 124.94, 125.02, 125.10, 126.34, 126.46, 127.81, 130.16, 131.03, 134.94, 135.52, 142.02, 147.70, 168.72, 192.70; ESI-MS m/z 628.0 (M + H)+.
(Z)-5-(2,6-Dichlorobenzylsulfonyl)-3-({3-[3-(4-methylpiperazin-1-yl)propyl]-4-oxo-4,5,6,7-tetrahydro-1H-indol-2-yl}methylene)indolin-2-one (24). Yield: 60%; 1H NMR (DMSO-d6) δ 13.67 (s, 1H), 11.52 (brs, 1H), 8.20 (s, 1H), 7.85 (s, 1H), 7.46–7.51 (m, 3H), 7.37–7.42 (m, 1H), 7.06 (d, J = 8.2 Hz, 1H), 4.88 (s, 2H), 2.91–3.03 (m, 4H), 2.22–2.44 (m, 10H), 2.01–2.14 (m, 7H), 1.70 (d, J = 6.8 Hz, 2H); 13C NMR (DMSO-d6) δ 21.22, 21.84, 21.97, 26.59, 37.59, 44.66, 51.70, 53.72, 56.01, 56.95, 108.78, 114.22, 118.13, 118.83, 124.91, 125.06, 126.31, 126.48, 127.81, 130.16, 130.96, 135.03, 135.51, 142.00, 143.92, 147.67, 168.72, 192.68; ESI-MS m/z 641.0 (M + H)+.
(Z)-Ethyl-3-(2-{[5-(2,6-dichlorobenzylsulfonyl)-2-oxoindolin-3-ylidene]methyl}-4-oxo-4,5,6,7-tetrahydro-1H-indol-3-yl)propanoate (8). To a solution of 7 (105.5 mg, 0.1840 mmol) in EtOH (2.0 mL) was added with SOCl2 (0.10 mL). The reaction mixture was heated at reflux for 12 h. The solution was concentrated under reduced pressure. The residue was added with H2O (5.0 mL) and heated. The solution was cooled to room temperature and the resultant solids were collected to give the desired 8 (101.8 mg, 0.1692 mmol) in 92% yield: 1H NMR (DMSO-d6) δ 13.68 (s, 1H), 11.53 (brs, 1H), 8.27 (d, J = 1.1 Hz, 1H), 7.95 (s, 1H), 7.46–7.53 (m, 3H), 7.34–7.42 (m, 1H), 7.06 (d, J = 8.2 Hz, 1H), 4.88 (s, 2H), 4.02 (q, J = 7.1 Hz, 2H), 3.25 (t, J = 7.4 Hz, 2H), 2.94 (t, J = 6.0 Hz, 2H), 2.61 (t, J = 7.4 Hz, 2H), 2.38–2.45 (m, 2H), 2.01–2.11 (m, 2H), 1.13 (t, J = 7.1 Hz, 3H); 13C NMR (DMSO-d6) δ 13.13, 19.12, 21.84, 21.88, 33.49, 37.47, 56.90, 58.83, 108.81, 114.87, 118.32, 118.77, 124.93, 124.99, 125.34, 126.23, 126.63, 127.81, 130.14, 131.08, 132.42, 135.50, 142.11, 147.61, 168.68, 171.27, 192.87; ESI-MS m/z 601.0 (M + H)+.
(Z)-3-(2-{[5-(2,6-Dichlorobenzylsulfonyl)-2-oxoindolin-3-ylidene]methyl}-4-oxo-4,5,6,7-tetrahydro-1H-indol-3-yl)-N-methylpropanamide (9). Yield: 85%; 1H NMR (DMSO-d6) δ 13.61 (s, 1H), 11.49 (brs, 1H), 8.22 (d, J = 1.6 Hz, 1H), 7.90 (s, 1H), 7.72 (dd, J = 4.2, 4.2 Hz, 1H), 7.46–7.53 (m, 3H), 7.39 (dd, J = 8.7, 7.4 Hz, 1H), 7.06 (d, J = 8.2 Hz, 1H), 4.88 (s, 2H), 3.22 (t, J = 7.3 Hz, 2H), 2.94 (t, J = 6.1 Hz, 2H), 2.48 (s, 3H), 2.45–2.40 (m, 2H), 2.37 (t, J = 7.3 Hz, 2H), 2.03–2.10 (m, 2H); 13C NMR (DMSO-d6) δ 19.67, 21.85, 21.93, 24.44, 35.34, 37.54, 56.90, 108.80, 114.46, 118.06, 118.74, 125.00, 125.37, 126.23, 126.55, 127.81, 130.18, 131.10, 133.66, 135.51, 142.06, 147.57, 168.67, 170.75, 192.81; ESI-MS m/z 586.0 (M + H)+.
(Z)-3-(2-{[5-(2,6-Dichlorobenzylsulfonyl)-2-oxoindolin-3-ylidene]methyl}-4-oxo-4,5,6,7-tetrahydro-1H-indol-3-yl)-N-[2-(diethylamino)ethyl]propanamide (10). Yield: 72%; 1H NMR (DMSO-d6) δ 13.60 (s, 1H), 11.49 (brs, 1H), 8.19 (s, 1H), 7.89 (s, 1H), 7.64 (d, J = 5.5 Hz, 1H), 7.46–7.54 (m, 3H), 7.35–7.43 (m, 1H), 7.06 (d, J = 8.2 Hz, 1H), 4.87 (s, 2H), 3.18–3.27 (m, 2H), 2.89–3.03 (m, 4H), 2.40–2.45 (m, 2H), 2.37 (t, J = 7.2 Hz, 2H), 2.28 (q, J = 7.0 Hz, 4H), 2.22 (t, J = 7.2 Hz, 2H), 2.01–2.12 (m, 2H), 0.77 (t, J = 7.0 Hz, 6H); 13C NMR (DMSO-d6) δ 10.56, 19.74, 21.85, 21.93, 35.47, 35.93, 37.55, 45.51, 50.64, 56.89, 108.76, 114.41, 117.94, 118.73, 124.93, 125.05, 125.49, 126.34, 126.47, 127.80, 130.17, 131.13, 133.50, 135.52, 142.07, 147.50, 168.69, 170.21, 192.79; ESI-MS m/z 671.0 (M + H)+.
(Z)-5-(2,6-Dichlorobenzylsulfonyl)-3-({4-oxo-3-[3-oxo-3-(pyrrolidin-1-yl)propyl]-4,5,6,7-tetrahydro-1H-indol-2-yl}methylene)indolin-2-one (11). Yield: 82%; 1H NMR (DMSO-d6) δ 13.64 (s, 1H), 11.44 (s, 1H), 8.22 (d, J = 1.3 Hz, 1H), 7.94 (s, 1H), 7.46–7.51 (m, 3H), 7.35–7.42 (m, 1H), 7.05 (d, J = 8.2 Hz, 1H), 4.88 (s, 2H), 3.19–3.39 (m, 6H), 2.94 (t, J = 5.9 Hz, 2H), 2.52–2.57 (m, 2H), 2.40–2.45 (m, 2H), 2.03–2.09 (m, 2H), 1.79–1.86 (m, 2H), 1.68–1.76 (m, 2H); 13C NMR (DMSO-d6) δ 19.35, 21.85, 21.93, 22.97, 24.65, 34.28, 37.54, 44.32, 44.83, 56.86, 108.75, 114.49, 118.19, 118.85, 124.99, 125.06, 125.41, 126.19, 126.48, 127.82, 130.15, 131.04, 133.81, 135.51, 142.04, 147.58, 168.70, 168.75, 192.86; ESI-MS m/z 626.0 (M + H)+.
(Z)-5-(2,6-Dichlorobenzylsulfonyl)-3-({3-[3-(4-methylpiperazin-1-yl)-3-oxopropyl]-4-oxo-4,5,6,7-tetrahydro-1H-indol-2-yl}methylene)indolin-2-one (12). Yield: 70%; 1H NMR (DMSO-d6) δ 13.66 (s, 1H), 11.51 (brs, 1H), 8.21 (d, J = 0.93 Hz, 1H), 7.91 (s, 1H), 7.47–7.50 (m, 3H), 7.35–7.42 (m, 1H), 7.05 (d, J = 8.2 Hz, 1H), 4.88 (s, 2H), 3.37–3.45 (m, 4H), 3.20 (t, J = 7.6 Hz, 2H), 2.94 (t, J = 6.0 Hz, 2H), 2.57 (t, J = 7.6 Hz, 2H), 2.43 (t, J = 6.0 Hz, 2H), 2.14–2.17 (m, 4H), 2.12 (s, 3H), 2.04–2.09 (m, 2H); 13C NMR (DMSO-d6) δ 19.85, 21.86, 21.93, 32.63, 37.53, 39.90, 43.75, 44.65, 53.31, 53.76, 56.89, 108.76, 114.63, 118.27, 118.89, 124.99, 125.39, 126.23, 126.54, 127.83, 130.17, 131.05, 133.42, 135.51, 142.06, 147.62, 168.70, 169.02, 192.98; ESI-MS m/z 655.0 (M + H)+.
Biological and computational methods
Acknowledgements
We are grateful to the Ministry of Economic Affairs of the Republic of China and Ministry of Science and Technology of Republic of China (102-2113-M-415-005-MY2) for financial support.Notes and references
- R. Haddad, K. E. Lipson and C. P. Webb, Anticancer Res., 2001, 21, 4243–4252 CAS.
- P. Longati, P. M. Comoglio and A. Bardelli, Curr. Drug Targets, 2001, 2, 41–55 CrossRef CAS.
- G. Maulik, A. Shrikhande, T. Kijima, P. C. Ma, P. T. Morrison and R. Salgia, Cytokine Growth Factor Rev., 2002, 13, 41–59 CrossRef CAS.
- S. Giordano, C. Ponzetto, M. F. Di Renzo, C. S. Cooper and P. M. Comoglio, Nature, 1989, 339, 155–156 CrossRef CAS PubMed.
- C. Birchmeier, W. Birchmeier, E. Gherardi and G. F. Vande Woude, Nat. Rev. Mol. Cell Biol., 2003, 4, 915–925 CrossRef CAS PubMed.
- C. M. Stellrecht and V. Gandhi, Cancer Lett., 2009, 280, 1–14 CrossRef CAS PubMed.
- X. Liu, R. C. Newton and P. A. Scherle, Trends Mol. Med., 2009, 16, 37–45 CrossRef PubMed.
- P. M. Comoglio, S. Giordano and L. Trusolino, Nat. Rev. Drug Discovery, 2008, 7, 504–516 CrossRef CAS PubMed.
- M. Sattler, Y. B. Pride, P. Ma, J. L. Gramlich, S. C. Chu, L. A. Quinnan, S. Shirazian, C. Liang, K. Podar, J. G. Christensen and R. Salgia, Cancer Res., 2003, 63, 5462–5469 CAS.
- J. G. Christensen, R. Schreck, J. Burrows, P. Kuruganti, E. Chan, P. Le, J. Chen, X. Wang, L. Ruslim, R. Blake, K. F. Lipson, J. Ramphal, S. Do, J. J. Cui, J. M. Cherrington and D. B. Mendel, Cancer Res., 2003, 63, 7345–7355 CAS.
- X. Wang, P. Le, C. Liang, J. Chan, D. Kiewlich, T. Miller, D. Harris, L. Sun, A. Rice, S. Vasile, R. A. Blake, A. R. Howlett, N. Patel, G. McMahon and K. E. Lipson, Mol. Cancer Ther., 2003, 2, 1085–1092 CAS.
- S. F. Bellon, P. Kaplan-Lefko, Y. Yang, Y. Zhang, J. Moriguchi, K. Rex, C. W. Johnson, P. E. Rose, A. M. Long, A. B. O'Connor, Y. Gu, A. Coxon, T. S. Kim, A. Tasker, T. L. Burgess and I. Dussault, J. Biol. Chem., 2008, 283, 2675–2683 CrossRef CAS PubMed.
- H. Y. Zou, Q. Li, J. H. Lee, M. E. Arango, S. R. McDonnell, S. Yamazaki, T. B. Koudriakova, G. Alton, J. J. Cui, P.-P. Kung, M. D. Nambu, G. Los, S. L. Bender, B. Mroczkowski and J. G. Christensen, Cancer Res., 2007, 67, 4408–4417 CrossRef CAS PubMed.
- M. Krug and A. Hilgeroth, Mini-Rev. Med. Chem., 2008, 8, 1312–1327 CrossRef CAS.
- L. Sun, N. Tran, C. Liang, F. Tang, A. Rice, R. Schreck, K. Waltz, L. K. Shawver, G. McMahon and C. Tang, J. Med. Chem., 1999, 42, 5120–5130 CrossRef CAS PubMed.
- J.-J. Huang, C.-C. Chiang, C. Liu, C.-L. Lai, S. F. Lin, Y.-H. Lin, R.-W. Wang, S.-c. Yang, J.-M. Chang, S.-H. Chuang and P. Chen, US Pat., 7897602, March 1, 2011.
- C.-C. Chiang, Y.-H. Lin, S. F. Lin, C.-L. Lai, C. Liu, W.-Y. Wei, S.-c. Yang, R.-W. Wang, L.-W. Teng, S.-H. Chuang, J.-M. Chang, T.-T. Yuan, Y.-S. Lee, P. Chen, W.-K. Chi, J.-Y. Yang, H.-J. Huang, C.-B. Liao and J.-J. Huang, J. Med. Chem., 2010, 53, 5929–5941 CrossRef CAS PubMed.
- G. Gambarotta, C. Boccaccio, S. Giordano, M. Andŏ, M. C. Stella and P. M. Comoglio, Oncogene, 1996, 13, 1911–1917 CAS.
- G. A. Smolen, R. Sordella, B. Muir, G. Mohapatra, A. Barmettler, H. Archibald, W. J. Kim, R. A. Okimoto, D. W. Bell, D. C. Sgroi, J. G. Christensen, J. Settleman and D. A. Haber, Proc. Natl. Acad. Sci., 2006, 103, 2316–2321 CrossRef CAS PubMed.
- M. A. Fabian, W. H. Biggs III, D. K. Treiber, C. E. Atteridge, M. D. Azimioara, M. G. Benedetti, T. A. Carter, P. Ciceri, P. T. Edeen, M. Floyd, J. M. Ford, M. Galvin, J. L. Gerlack, R. M. Grotzfeld, S. Herrgard, D. E. Insko, M. A. Insko, A. G. Lai, J.-M. Lelias, S. A. Mehta, Z. V. Milanov, A. M. Velasco, L. M. Wodicka, H. K. Patel, P. P. Zarrinkar and D. J. Lockhart, Nat. Biotechnol., 2005, 23, 329–336 CrossRef CAS PubMed.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng., 1995, 8, 127–134 CrossRef CAS PubMed.
- L. Sun, N. Tran, F. Tang, H. App, P. Hirth, G. McMahon and C. Tang, J. Med. Chem., 1998, 41, 2588–2603 CrossRef CAS PubMed.
- H. Guan, A. D. Laird, R. A. Blake, C. Tang and C. Liang, Bioorg. Med. Chem. Lett., 2004, 14, 187–190 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Ligplot diagrams of the ATP binding site of Met complexed with compounds 2 and 20, and the kinase profiling data of 20. See DOI: 10.1039/c4ra08720h |
| This journal is © The Royal Society of Chemistry 2014 |