
Study of the post separation pH adjustment by a microchip for the analysis of aminoglycoside antibiotics†
Abstract
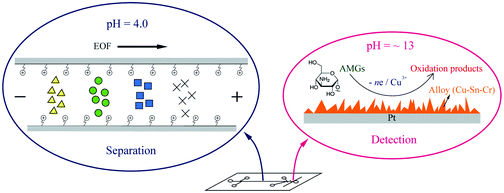
Recently, we developed a simple microchip to simultaneously accommodate acidic conditions for the separation and alkaline conditions for the electrochemical detection of aminoglycoside antibiotics [Electrophoresis, DOI: 10.1002/elps.201200309]. With two branch channels connected near the end of the separation channel, the alkaline solution was hydrostatically introduced into a Z-shaped mixing channel and combined with the acidic stream from the separation channel. As a result, the pH of the mixed solution was adjusted to the desired value for the electrochemical detection of aminoglycoside antibiotics. In this manuscript, the principle and related parameters of the pH adjustment on the microchip were investigated both in theory and practice. With the guidance of the principle of the post separation pH adjustment, we applied the functional microchip to the analysis of six aminoglycoside antibiotics in a biological sample with satisfactory analytical performances. Specifically, these compounds were electrophoretically separated in 5 mM sodium acetate (pH = 4) and 0.6 mM CTAB, and through the post separation pH adjustment, electrochemically detected in alkaline conditions (pH > 12) at a Cu–Sn–Cr alloy electrode. Additionally, this microchip may provide a possible use for the post separation reagent addition for enzyme-assisted electrochemical detection.
 Please wait while we load your content...
Please wait while we load your content...

