
Tuning the band gap and optical properties of γ-Si3N4 with rare earth element Ce
Abstract
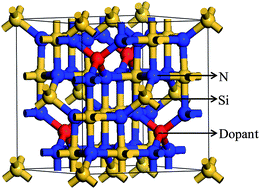
The present paper reports the investigation of optoelectronic properties of doped γ-Si3N4. The rare earth element Ce is used as a dopant. Two structures are picked out from the four ones with different doping methods by comparing their formation and binding energies. The effects of doping methods and concentrations are explored based on the favourable structures. Four cases with different concentrations are considered for each doping method. Their electronic and optical properties are calculated on the basis of hybrid density functional theory with Hubbard U correction. The Ce dopant with the proper doping method and concentration results in decreased band gaps, improved low-energy electron transition and enhanced absorption of solar energy. The mechanism for the doping effect is elucidated by analyzing the electronic structure.
 Please wait while we load your content...
Please wait while we load your content...

